Sub-sampling the data of a toffee file to just include standard peptides¶
In order to build a set of test files with known properties that can be used within a testing framework, we wish to take a full experimental file and extract out a subset of the data. In this example, we are going to take a HEK293 control that has a collection of spike-in retention time standards, extract the data just for those peptides such that this file can be used in regression tests.
[1]:
%load_ext autoreload
%autoreload 2
[2]:
%matplotlib inline
import os
import pandas as pd
import numpy as np
import scipy
import scipy.sparse
import seaborn as sns
import matplotlib.pyplot as plt
from tqdm import tqdm
import toffee
from toffee import util
from toffee.manipulation import Subsampler
sns.set()
sns.set_color_codes()
[3]:
toffee.__version__
[3]:
'0.12.18'
Sub-sample using the Spectral Reference Library (SRL)¶
Open the transition library for the iRT peptides
[4]:
base_dir = os.environ.get('DIA_TEST_DATA_REPO', None)
assert base_dir is not None
srl_fname = base_dir + '/ProCan90/srl/hek_srl.OpenSwath.iRT.tsv'
srl = pd.read_csv(srl_fname, sep='\t')
and then create the object for sub-sampling a toffee file using the peptides in teh reference library.
[5]:
tof_fname = base_dir + '/ProCan90/tof/ProCan90-M03-01.tof'
subsampler = Subsampler(
tof_fname=tof_fname,
precursor_and_product_df=srl,
filter_ms2_windows=['ms2-025', 'ms2-041'],
)
Running the sub-sampling is as simple as passing in the output file name.
[6]:
test_tof_fname = os.path.basename(tof_fname).replace('.tof', '.test.tof')
subsampler.run(test_tof_fname)
100%|██████████| 2/2 [00:25<00:00, 11.43s/it]
Properties of the toffee HDF5 file¶
Using standard HDF tools we can interrogate the new toffee file for things such as compression, dataset and attribute names, or even the Intrinsic Mass Spacing parameters.
[7]:
!ls -lhF $test_tof_fname
-rw-r--r-- 1 btully 470603294 867K 15 Apr 20:43 ProCan90-M03-01.test.tof
[8]:
!h5ls -vg $test_tof_fname/ms1
Opened "ProCan90-M03-01.test.tof" with sec2 driver.
ms1 Group
Attribute: IMSAlpha scalar
Type: native double
Data: 7.01548e-05
Attribute: IMSBeta scalar
Type: native double
Data: -5.92871e-05
Attribute: IMSGamma scalar
Type: native int
Data: 266664
Attribute: firstScanRetentionTimeOffset scalar
Type: native double
Data: 0.17
Attribute: precursorCenter scalar
Type: native double
Data: -1
Attribute: precursorLower scalar
Type: native double
Data: -1
Attribute: precursorUpper scalar
Type: native double
Data: -1
Attribute: scanCycleTime scalar
Type: native double
Data: 3.32
Location: 1:405261
Links: 1
[9]:
!h5ls -v $test_tof_fname/ms1
Opened "ProCan90-M03-01.test.tof" with sec2 driver.
IMSAlphaPerScan Dataset {1627/1627}
Location: 1:413217
Links: 1
Chunks: {1627} 13016 bytes
Storage: 13016 logical bytes, 6975 allocated bytes, 186.61% utilization
Filter-0: deflate-1 OPT {6}
Type: native double
IMSBetaPerScan Dataset {1627/1627}
Location: 1:422560
Links: 1
Chunks: {1627} 13016 bytes
Storage: 13016 logical bytes, 7470 allocated bytes, 174.24% utilization
Filter-0: deflate-1 OPT {6}
Type: native double
imsCoord Dataset {257400/257400}
Location: 1:432398
Links: 1
Chunks: {257400} 1029600 bytes
Storage: 1029600 logical bytes, 198728 allocated bytes, 518.10% utilization
Filter-0: deflate-1 OPT {6}
Type: native unsigned int
intensity Dataset {257400/257400}
Location: 1:633670
Links: 1
Chunks: {257400} 1029600 bytes
Storage: 1029600 logical bytes, 251727 allocated bytes, 409.01% utilization
Filter-0: deflate-1 OPT {6}
Type: native unsigned int
retentionTimeIdx Dataset {1627/1627}
Location: 1:406565
Links: 1
Chunks: {1627} 6508 bytes
Storage: 6508 logical bytes, 3956 allocated bytes, 164.51% utilization
Filter-0: deflate-1 OPT {6}
Type: native unsigned int
[10]:
!h5ls -vg $test_tof_fname/ms2-041
Opened "ProCan90-M03-01.test.tof" with sec2 driver.
ms2-041 Group
Attribute: IMSAlpha scalar
Type: native double
Data: 7.01547e-05
Attribute: IMSBeta scalar
Type: native double
Data: -3.32516e-05
Attribute: IMSGamma scalar
Type: native int
Data: 142548
Attribute: firstScanRetentionTimeOffset scalar
Type: native double
Data: 1.467
Attribute: precursorCenter scalar
Type: native double
Data: 611.5
Attribute: precursorLower scalar
Type: native double
Data: 608.5
Attribute: precursorUpper scalar
Type: native double
Data: 614.5
Attribute: scanCycleTime scalar
Type: native double
Data: 3.32
Location: 1:174499
Links: 1
[11]:
!h5ls -v $test_tof_fname/ms2-041
Opened "ProCan90-M03-01.test.tof" with sec2 driver.
IMSAlphaPerScan Dataset {1627/1627}
Location: 1:181206
Links: 1
Chunks: {1627} 13016 bytes
Storage: 13016 logical bytes, 8244 allocated bytes, 157.88% utilization
Filter-0: deflate-1 OPT {6}
Type: native double
IMSBetaPerScan Dataset {1627/1627}
Location: 1:191818
Links: 1
Chunks: {1627} 13016 bytes
Storage: 13016 logical bytes, 4755 allocated bytes, 273.73% utilization
Filter-0: deflate-1 OPT {6}
Type: native double
imsCoord Dataset {96051/96051}
Location: 1:198941
Links: 1
Chunks: {96051} 384204 bytes
Storage: 384204 logical bytes, 172435 allocated bytes, 222.81% utilization
Filter-0: deflate-1 OPT {6}
Type: native unsigned int
intensity Dataset {96051/96051}
Location: 1:373920
Links: 1
Chunks: {96051} 384204 bytes
Storage: 384204 logical bytes, 28973 allocated bytes, 1326.08% utilization
Filter-0: deflate-1 OPT {6}
Type: native unsigned int
retentionTimeIdx Dataset {1627/1627}
Location: 1:175803
Links: 1
Chunks: {1627} 6508 bytes
Storage: 6508 logical bytes, 2707 allocated bytes, 240.41% utilization
Filter-0: deflate-1 OPT {6}
Type: native unsigned int
Compare to the original data¶
By opening up the file we just created, we can ensure that the data it contains matches exactly the same data as the original file. In doing so, we can prove that the data used in testing later is indistiguisable regardless of which file is being used.
First off, we need a series of functions that check data inside numpy and scipy matrices.
[12]:
def sparse_assert_allclose(A, B, atol=1e-8):
"""
Test to see if two sparse matrices compare equal.
Modified from: https://stackoverflow.com/a/47771340/758811
"""
np.testing.assert_array_equal(A.shape, B.shape)
r1, c1, v1 = scipy.sparse.find(A)
r2, c2, v2 = scipy.sparse.find(B)
np.testing.assert_array_equal(r1, r2)
np.testing.assert_array_equal(c1, c2)
np.testing.assert_allclose(v1, v2, atol=atol)
def chromatogram_assert_allclose(A, B):
np.testing.assert_allclose(
A.retentionTime,
B.retentionTime
)
np.testing.assert_allclose(
A.massOverCharge,
B.massOverCharge
)
sparse_assert_allclose(
A.intensities,
B.intensities
)
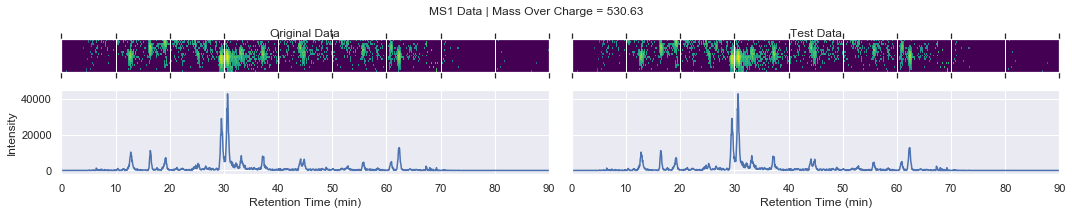
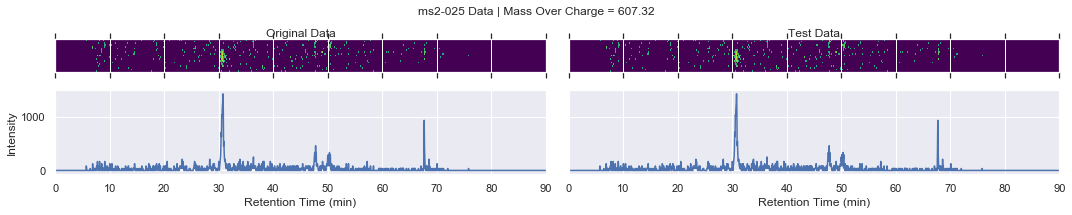
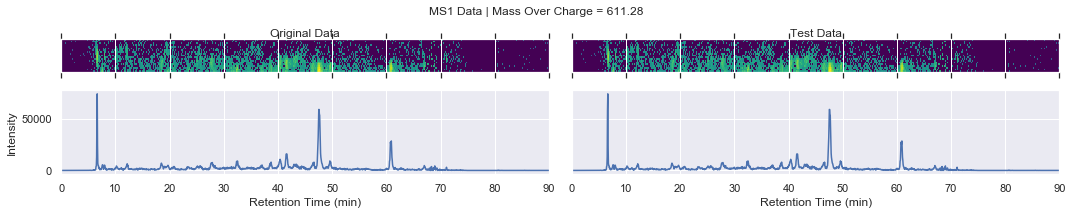
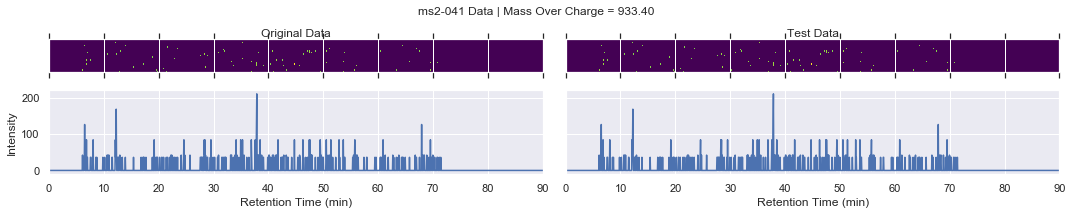
For further checking, we can plot the raw data of each file alongside one another
[13]:
def plot_raw(original_chrom, test_chrom, mz_value, window):
def _plot_raw_impl(chrom, ax1, ax2, title, label_y=True):
xlim = [chrom.retentionTime[0] / 60.0, chrom.retentionTime[-1] / 60.0]
dense_intensities = chrom.intensities.toarray()
intensities = np.log10(1 + dense_intensities.T)
ax1.matshow(intensities, extent=xlim + [0, 6.0], cmap=plt.cm.viridis)
ax1.set_xlim(xlim)
ax1.set_ylabel('m/z (Da)')
ax1.yaxis.set_visible(False)
ax1.set_title(title, y=0.9)
ax2.plot(chrom.retentionTime / 60.0, np.sum(dense_intensities, axis=1))
ax2.set_xlim(xlim)
ax2.set_xlabel('Retention Time (min)')
if label_y:
ax2.set_ylabel('Intensity')
fig, axes = plt.subplots(nrows=2, ncols=2, figsize=(15, 3), sharex=True, sharey='row')
_plot_raw_impl(original_chrom, axes[0, 0], axes[1, 0], 'Original Data')
_plot_raw_impl(test_chrom, axes[0, 1], axes[1, 1], 'Test Data', label_y=False)
fig.suptitle('{} Data | Mass Over Charge = {:.2F}'.format(window, mz_value))
fig.tight_layout()
plt.show()
Again, loading the MS1 data is more expensive than others, so we only want to do it once
[14]:
test_swath_run = toffee.SwathRun(test_tof_fname)
test_ms1_swath_map = test_swath_run.loadSwathMap(toffee.ToffeeWriter.MS1_NAME)
Finally, let’s loop through the precursors and products that were used by the sub-samplerand compare the new file to the existing data
[15]:
ppm_width = 100
for k, ms2_name in enumerate(tqdm(sorted(subsampler.precursors.ms2Name.unique().tolist()))):
# filter the library
try:
precursors = subsampler.precursors[subsampler.precursors.ms2Name == ms2_name]
products = subsampler.products[subsampler.products.ms2Name == ms2_name]
except KeyError:
# this will happen if the window is not in the library
continue
# load the MS2 swath map
ms2_swath_map = subsampler.swath_run.loadSwathMap(ms2_name)
test_ms2_swath_map = test_swath_run.loadSwathMap(ms2_name)
# collect the MS1 data
for i, (_, row) in enumerate(precursors.iterrows()):
mz = row.IsotopeMz
mz_range = toffee.MassOverChargeRangeWithPPMFullWidth(mz, ppm_width)
ms1_xic = subsampler.ms1_swath_map.extractedIonChromatogramSparse(mz_range)
test_ms1_xic = test_ms1_swath_map.extractedIonChromatogramSparse(mz_range)
if i == 0 and k < 4:
plot_raw(ms1_xic, test_ms1_xic, mz, window='MS1')
try:
chromatogram_assert_allclose(ms1_xic, test_ms1_xic)
except AssertionError:
plot_raw(ms1_xic, test_ms1_xic, mz, window=f'Error! MS1')
# collect the MS2 data
for i, (_, row) in enumerate(products.iterrows()):
mz = row.IsotopeMz
mz_range = toffee.MassOverChargeRangeWithPPMFullWidth(mz, ppm_width)
ms2_xic = ms2_swath_map.extractedIonChromatogramSparse(mz_range)
test_ms2_xic = test_ms2_swath_map.extractedIonChromatogramSparse(mz_range)
if i == 0 and k < 4:
plot_raw(ms2_xic, test_ms2_xic, mz, window=ms2_name)
try:
chromatogram_assert_allclose(ms2_xic, test_ms2_xic)
except AssertionError:
plot_raw(ms2_xic, test_ms2_xic, mz, window=f'Error! {ms2_name}')
0%| | 0/2 [00:00<?, ?it/s]
50%|█████ | 1/2 [00:01<00:01, 1.96s/it]
100%|██████████| 2/2 [00:03<00:00, 1.82s/it]
And now, clean up
[16]:
if os.path.isfile(test_tof_fname):
os.remove(test_tof_fname)