Re-Quantifying detections using toffee and 2D modified Gaussian¶
Through the many visualisations of toffee data, and appreciation for the TOF detector, one recognises that the data is roughly gaussian in both retention time and m/z index space. Indeed, as we will see below, it is possible to model the raw data as an analytic function to improve the quantification results. In doing so, the least-squares method used to fit the function to the data mimicks a high-frequency filter and performs basic noise removal. By fixing the retention time of the analytic function for all fragments, it is also possible to pull apart co-eluting peaks and count only the contribution from the peak of interest.
The elution profile from the LC column gives a log-normal distribution that is skewed towards the left, while the data is normally distributed in index m/z space that we attribute to the distribution of kinetic energy that individual ions obtain within the TOF mass analyser. These observations imply that we can perform an optimization to find the peak location, spread, and amplitude of the gaussian functions for each fragment, \(j\).
with chemical noise, for MS1 signal only, defined as
and the minimisation function
where - \(t\) and \(m\) represent the retention time and \(i_\sqrt{m/z}\) space respectively - \(t_0\) and \(\vec{m_0}\) are the peak location, where we assume that the retention time for all fragments must be constant, but the mass offset is allowed to be different for each one to account for calibration offsets - \(\sigma_t\), \(\sigma_m\) are the spread of each gaussian - \(\vec{a}\) is the amplitude for each fragment - \(\vec{c}\) is the amplitude of chemical noise for each fragment, and - \(I_j\) is the raw intensity data for a given fragment.
By building a model such as a 2D gaussian, we are then able to calculate the volume under the curve, with a hypothesis that this should give a more robust measure of the intensity from sample to sample.
Furthermore, to improve robustness during least-squares fitting, we apply the following transforms to the parameters:
\(f(n, x) = n (1 + \tanh{x}) / 2\) is a sigmoid function for constraining \(x\) to the range of \(0, n\)
\(\sigma_t = \sigma_t^{\prime2}\), where \(\sigma_t^\prime\) is the value used during optimisation
\(\sigma_m = \sigma_m^{\prime2}\), where \(\sigma_m^\prime\) is the value used during optimisation
\(t_0 = f(num_t, t_0^\prime)\), where \(t_0^\prime\) is the value used during optimisation, and \(num_t\) is the number of retention time pixels. This constrains \(t_0\) to fall within the valid range of retention times
\(m_0 = f(num_m, m_0^\prime)\), where \(m_0^\prime\) is the value used during optimisation, and \(num_m\) is the number of index m/z pixels. This constrains \(m_0\) to fall within the valid range of index m/z values
\(a_j = a_j^{\prime2}\), where \(a_j^\prime\) is the value used during optimisation
\(c_j = c_j^{\prime2}\), where \(c_j^\prime\) is the value used during optimisation
[1]:
%matplotlib inline
%load_ext autoreload
%autoreload 2
import os
import datetime
from IPython.display import SVG, Image, display
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import seaborn as sns
import toffee
from toffee import requant
from toffee.util import calculate_isotope_mz
from toffee.viz import ToffeeFragmentsPlotter
from tqdm import tqdm
base_dir = os.environ.get('DIA_TEST_DATA_REPO', None)
assert base_dir is not None
sns.set()
[2]:
print(' Analysis date:', datetime.datetime.now())
print('toffee version:', toffee.__version__)
Analysis date: 2019-08-12 10:15:31.826510
toffee version: 0.14.2
Fitting the data to an analytic model using the C++ library¶
In toffee.requant there are two classes that enable the requantification to be performed using PyProphet results as the input.
Swath Gold Standard data¶
[3]:
sgs_files = [2, 5, 8, 10]
force = False
sgs_quantifiers = {}
sgs_requantified = []
for f in tqdm(sgs_files):
name = f'{f:03d}'
sample = f'napedro_l120224_{name}_sw'
output = sample + '.csv.gz'
tof_fname = f'{base_dir}/SwathGoldStandard/tof/{sample}.tof'
pyprophet_fname = f'{base_dir}/SwathGoldStandard/osw/{sample}.osw'
q = requant.PyProphetSQLiteRequantifier(
output_filename=output,
toffee_filename=tof_fname,
pyprophet_filename=pyprophet_fname,
max_protein_q_value_rs=1.0,
max_protein_q_value_experiment_wide=1.0,
max_protein_q_value_global=1.0,
)
sgs_quantifiers[f] = q
if not os.path.exists(output) or force:
q.run()
df = pd.read_csv(output)
df['InjectionName'] = name
df['PQM'] = df.FullPeptideName + '_' + df.Charge.map(str)
sgs_requantified.append(df)
sgs_requantified = pd.concat(sgs_requantified)
sgs_requantified.head()
100%|██████████| 4/4 [00:00<00:00, 10.48it/s]
[3]:
| run_id | peptide_id | Sequence | FullPeptideName | Charge | peak_group_rank | RT | Intensity | m_score | m_score_peptide_run_specific | ... | MS1Intensity | MS2Intensity | ModelParamSigmaRT | ModelParamSigmaMz | ModelParamRT0 | ModelParamMz0MS1 | ModelParamMz0MS2 | ModelParamAmplitudes | InjectionName | PQM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1368487973123354311 | 1 | AAEDFTLLVK | AAEDFTLLVK(UniMod:259) | 2 | 1 | 4209.56 | 80.0 | 0.032005 | 0.032005 | ... | 5.028338 | 1.668722 | 0.064002 | 11.891527 | 15.757658 | 14.731366 | 12.945118 | 8.66231,8.142178.14217;1.04211.0421,2.753782.7... | 002 | AAEDFTLLVK(UniMod:259)_2 |
| 1 | 1368487973123354311 | 4 | AAGASAQVLGQEGK | AAGASAQVLGQEGK(UniMod:259) | 2 | 1 | 2017.42 | 822.0 | 0.003899 | 0.003899 | ... | 76.928972 | 15.230757 | 0.088230 | 5.231933 | 14.949945 | 20.636998 | 20.908421 | 305.804,170.775170.775;41.441141.4411,16.10781... | 002 | AAGASAQVLGQEGK(UniMod:259)_2 |
| 2 | 1368487973123354311 | 17 | ADSTGTLVITDPTR | ADSTGTLVITDPTR(UniMod:267) | 2 | 1 | 3105.00 | 180.0 | 0.003899 | 0.003899 | ... | 8.074463 | 2.976146 | 0.065825 | 2.685659 | 15.040877 | 20.335423 | 21.117461 | 49.0696,42.791442.7914;2.926292.92629,8.41438.... | 002 | ADSTGTLVITDPTR(UniMod:267)_2 |
| 3 | 1368487973123354311 | 22 | AEVAALAAENK | AEVAALAAENK(UniMod:259) | 2 | 1 | 2221.28 | 2658.0 | 0.003899 | 0.003899 | ... | 189.337273 | 57.394183 | 0.082998 | 5.633617 | 15.131340 | 20.949316 | 21.025522 | 810.276,374.571374.571;70.898270.8982,87.69587... | 002 | AEVAALAAENK(UniMod:259)_2 |
| 4 | 1368487973123354311 | 26 | AFGYYGPLR | AFGYYGPLR(UniMod:267) | 2 | 1 | 3723.90 | 320.0 | 0.034796 | 0.034796 | ... | 19.167878 | 9.666694 | 0.110380 | 9.718412 | 14.489525 | 23.953597 | 19.389373 | 35.5049,38.795338.7953;8.423798.42379,7.632937... | 002 | AFGYYGPLR(UniMod:267)_2 |
5 rows × 24 columns
ProCan90 data¶
[4]:
procan90_files = ['03-01', '06-07']
force = False
procan90_quantifiers = {}
procan90_requantified = []
for f in tqdm(procan90_files):
sample = f'ProCan90-M{f}'
output = sample + '.csv.gz'
tof_fname = f'{base_dir}/ProCan90/tof/{sample}.tof'
pyprophet_fname = f'{base_dir}/ProCan90/osw/{sample}.osw'
q = requant.PyProphetSQLiteRequantifier(
output_filename=output,
toffee_filename=tof_fname,
pyprophet_filename=pyprophet_fname,
)
procan90_quantifiers[f] = q
if not os.path.exists(output) or force:
q.run()
df = pd.read_csv(output)
df['InjectionName'] = sample
df['PQM'] = df.FullPeptideName + '_' + df.Charge.map(str)
procan90_requantified.append(df)
procan90_requantified = pd.concat(procan90_requantified)
procan90_requantified.head()
100%|██████████| 2/2 [00:14<00:00, 7.43s/it]
[4]:
| run_id | peptide_id | Sequence | FullPeptideName | Charge | peak_group_rank | RT | Intensity | m_score | m_score_peptide_run_specific | ... | MS1Intensity | MS2Intensity | ModelParamSigmaRT | ModelParamSigmaMz | ModelParamRT0 | ModelParamMz0MS1 | ModelParamMz0MS2 | ModelParamAmplitudes | InjectionName | PQM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 5145023687975356512 | 0 | AAAAAAAAAAAAAAAGAGAGAK | AAAAAAAAAAAAAAAGAGAGAK | 3 | 1 | 2277.710 | 4944.0 | 0.016517 | 0.016516 | ... | 611.732180 | 88.886325 | 0.289517 | 3.981082 | 14.301614 | 19.422064 | 20.641688 | 929.679,4790.634790.63;198.689198.689,133.7331... | ProCan90-M03-01 | AAAAAAAAAAAAAAAGAGAGAK_3 |
| 1 | 5145023687975356512 | 3 | AAAAAAAAAAGRVGM | AAAAAAAAAAGRVGM | 2 | 1 | 1609.310 | 14029.0 | 0.000583 | 0.000583 | ... | 2086.219858 | 201.586612 | 0.218611 | 3.505546 | 15.515581 | 22.769187 | 21.015362 | 14288.9,6270.76270.7;757.188757.188,649649,387... | ProCan90-M03-01 | AAAAAAAAAAGRVGM_2 |
| 2 | 5145023687975356512 | 13 | AAAAAADPNAAWAAYYSHYYQQPPGPVPGPAPAPAAPPAQGEPPQP... | AAAAAADPNAAWAAYYSHYYQQPPGPVPGPAPAPAAPPAQGEPPQP... | 5 | 1 | 3275.670 | 2988.0 | 0.044867 | 0.044866 | ... | 289.512315 | 87.778535 | 0.291877 | 2.737636 | 13.559004 | 21.539127 | 21.589122 | 404.298,2301.282301.28;27.238927.2389,136.0631... | ProCan90-M03-01 | AAAAAADPNAAWAAYYSHYYQQPPGPVPGPAPAPAAPPAQGEPPQP... |
| 3 | 5145023687975356512 | 16 | AAAAADLANR | AAAAADLANR | 2 | 1 | 475.722 | 1992.0 | 0.012483 | 0.012482 | ... | 479.286636 | 134.605709 | 0.355179 | 11.511312 | 17.247381 | 19.503196 | 16.965990 | 545.168,1321.941321.94;221.035221.035,120.5912... | ProCan90-M03-01 | AAAAADLANR_2 |
| 4 | 5145023687975356512 | 19 | AAAAASAAGPGGLVAGK | AAAAASAAGPGGLVAGK | 2 | 1 | 978.743 | 978.0 | 0.004057 | 0.004057 | ... | 355.547847 | 31.256479 | 0.307980 | 7.227377 | 11.911672 | 9.497525 | 16.879286 | 1193.04,604.938604.938;17.968217.9682,66.87976... | ProCan90-M03-01 | AAAAASAAGPGGLVAGK_2 |
5 rows × 24 columns
Compare intensities calculated by the toffee.requant C++ library¶
[5]:
def plot_intensities(requantified, cols, colname, divisor=None, expected_intensity=None, ylim=None):
log_func = np.log2
log_func_name = '(log2)'
colname = f'{colname} {log_func_name}'
def load(df, col):
df = df[['PQM', 'InjectionName', col]].set_index(['PQM', 'InjectionName']).unstack('InjectionName')
if divisor is not None:
df = df.divide(df[(col, divisor)], axis=0)
df = df.apply(log_func)
return df.stack().reset_index(drop=False).rename(columns={col: colname})
results = []
for col in cols:
df = load(requantified, col)
df['Source'] = col
results.append(df)
results = pd.concat(results)
x = 'InjectionName'
col = 'Source'
if expected_intensity is not None:
x, col = col, x
g = sns.catplot(
kind='box',
data=results,
y=colname,
x=x,
col=col,
palette='viridis',
zorder=5,
sharey=ylim is not None,
showfliers=False,
)
if expected_intensity is not None:
for ax, e in zip(g.axes.flatten(), expected_intensity):
for i in range(len(results[x].unique())):
ax.plot([i - 0.5, i + 0.5], log_func([e, e]), 'r--', zorder=1)
if ylim is not None:
ax.set_ylim(log_func(ylim))
plt.show()
Swath Gold Standard data¶
[6]:
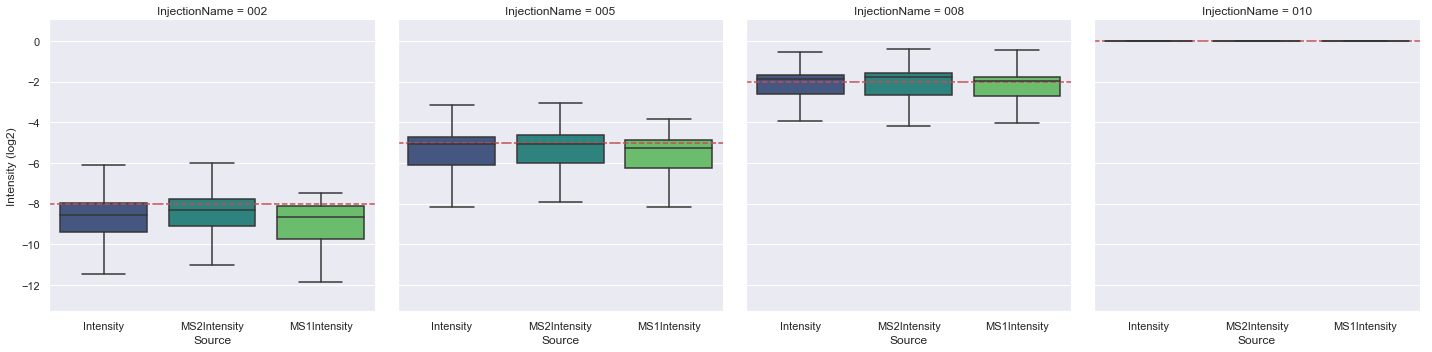
plot_intensities(
sgs_requantified,
['Intensity', 'MS2Intensity', 'MS1Intensity'],
'Intensity',
expected_intensity=[2.0 ** (d - 10) for d in sgs_files],
divisor='010',
ylim=(1e-4, 2),
)
ProCan90 data¶
[7]:
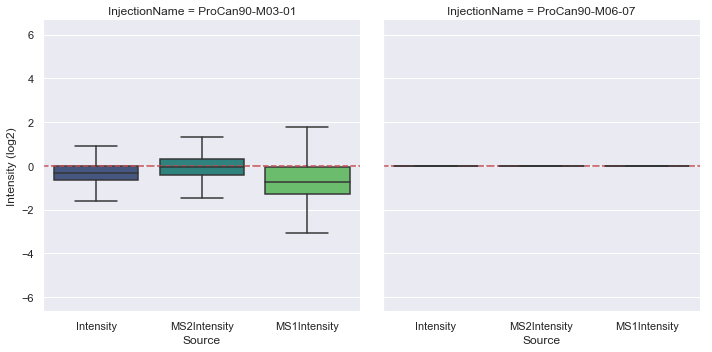
plot_intensities(
procan90_requantified,
['Intensity', 'MS2Intensity', 'MS1Intensity'],
'Intensity',
expected_intensity=(1.0, 1.0),
divisor='ProCan90-M06-07',
ylim=(0.01, 100),
)
Explore the raw data¶
High-scoring results¶
[8]:
def explore_raw_data(file, quantifiers, peptide_ids=None, plotter=None):
q = quantifiers[file]
if plotter is None:
plotter = ToffeeFragmentsPlotter(q.toffee_filename)
srl = q._add_ms2_windows(plotter.extractor.swath_run, q.srl)
if peptide_ids is None:
peptide_ids = q.scores \
.sort_values(['m_score', 'Intensity'], ascending=(True, False)) \
.iloc[100:115] \
.peptide_id \
.tolist()
all_srl = srl.loc[srl.peptide_id.isin(peptide_ids)].copy()
for ms2_name in sorted(all_srl.MS2Name.unique()):
print('*' * 80)
ms2_srl = all_srl.loc[all_srl.MS2Name == ms2_name]
for peptide_id in ms2_srl.peptide_id.unique():
print('-' * 50)
fragments = ms2_srl.loc[ms2_srl.peptide_id == peptide_id]
pre = fragments['PrecursorMz'].tolist()[:1]
pro = fragments['ProductMz'].tolist()[:6]
peptide = q.scores.loc[q.scores.peptide_id == peptide_id, 'FullPeptideName'].iloc[0]
charge = q.scores.loc[q.scores.peptide_id == peptide_id, 'Charge'].iloc[0]
rt = q.scores.loc[q.scores.peptide_id == peptide_id, 'RT'].iloc[0]
rt_zoom = (rt - 300.0, rt + 300.0)
print(f'ms2_name = \'{ms2_name}\'')
print(f'peptide_id, peptide, charge, rt, pre = \'{peptide_id}\', \'{peptide}\', {charge}, {rt}, {pre[0]}')
print(f'pro = {pro}')
output_fname = '/tmp/fig.png'
fig = plotter.create_plotly_figure(
precursor_mz_list=pre,
product_mz_list=pro,
psm_name='{} | extraction_width = {} {}'.format(
plotter.tof_fname.split('/')[-1].split('.')[0],
plotter.extractor.extraction_width,
'ppm' if plotter.extractor.use_ppm else 'px',
),
number_of_isotopes=0,
log_heatmap=True,
normalise_per_fragment=False,
retention_time_zoom=rt_zoom,
aspect_scale=0.2,
retention_time_line=rt,
output_fname=output_fname,
)
display(Image(output_fname))
SGS data¶
[9]:
sgs_file = 5
sgs_peptide_ids = [
476,
477,
723,
920,
]
explore_raw_data(
file=sgs_file,
quantifiers=sgs_quantifiers,
peptide_ids=sgs_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-008'
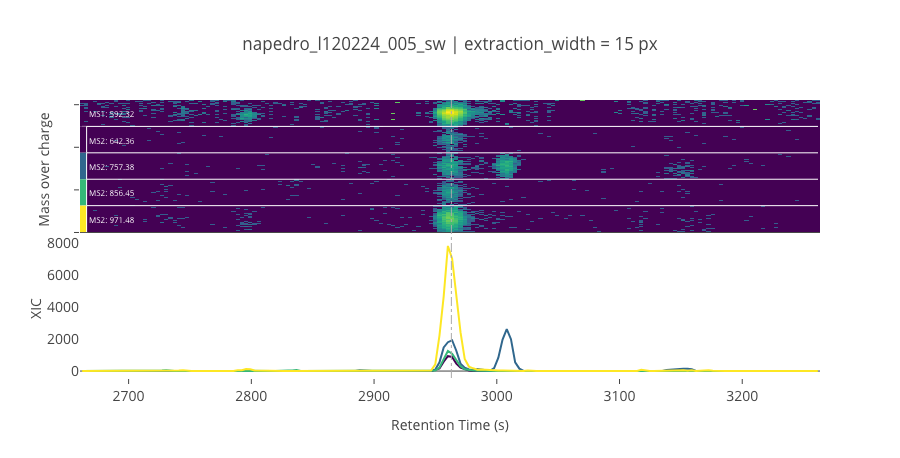
peptide_id, peptide, charge, rt, pre = '476', 'IVDVDLTSEGK(UniMod:259)', 2, 2962.99, 592.318
pro = [642.355, 757.382, 856.45, 971.477]
--------------------------------------------------
ms2_name = 'ms2-008'
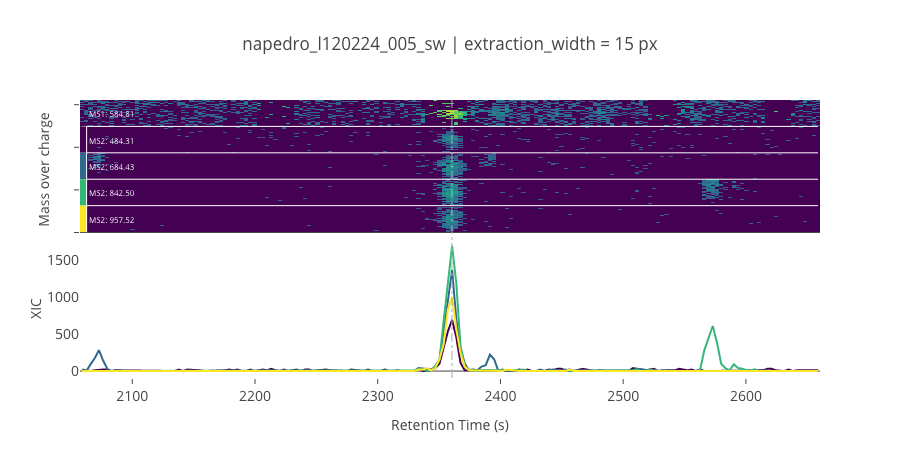
peptide_id, peptide, charge, rt, pre = '723', 'NPDGTVTVISR(UniMod:267)', 2, 2360.46, 584.813
pro = [484.312, 684.428, 842.497, 957.524]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-010'
peptide_id, peptide, charge, rt, pre = '477', 'IVEIYGPESSGK(UniMod:259)', 2, 2900.32, 643.84
pro = [669.329, 832.393, 945.477, 1074.52]
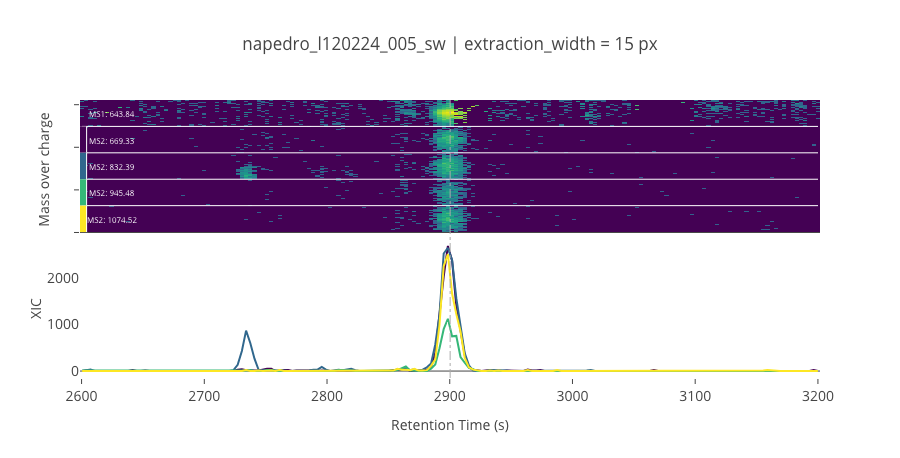
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-014'
peptide_id, peptide, charge, rt, pre = '920', 'SDSSDTPPLPSPPGK(UniMod:259)', 2, 2804.66, 745.367
pro = [800.476, 897.528, 998.576, 1113.6]
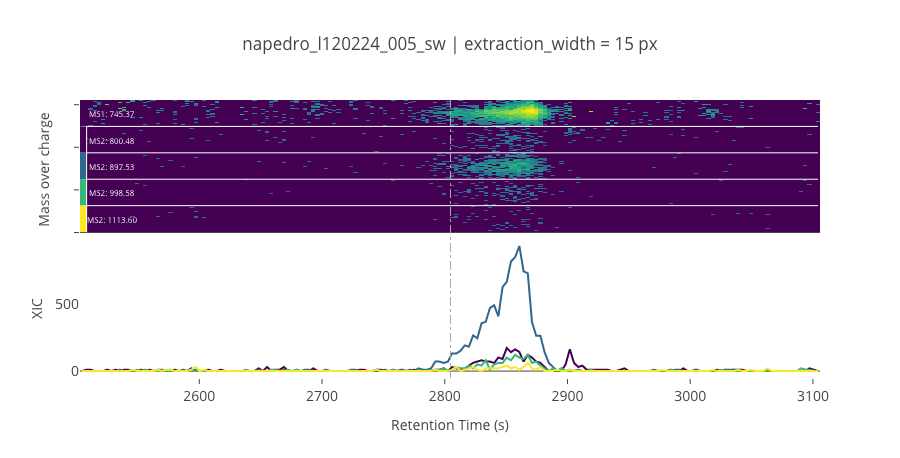
ProCan90 data¶
[10]:
procan90_file = '03-01'
procan90_peptide_ids = [ # these are found by examing the raw data
197043,
4114,
114292,
193918,
33530,
# 29585,
# 34385,
# 199044,
# 109281,
# 16535,
# 31437,
# 113336,
# 192320,
]
explore_raw_data(
file=procan90_file,
quantifiers=procan90_quantifiers,
peptide_ids=procan90_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-024'
peptide_id, peptide, charge, rt, pre = '197043', 'VSFELFADK', 2, 3025.44, 528.27404785
pro = [435.22381592, 480.24526978, 593.3293457, 722.37194824, 869.44036865, 956.47235107]
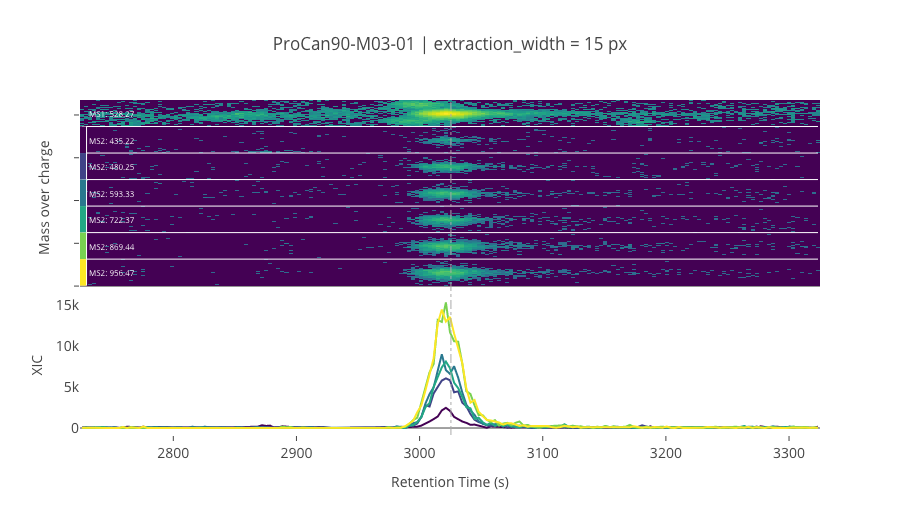
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-035'
peptide_id, peptide, charge, rt, pre = '4114', 'AIPQLQGYLR', 2, 2607.96, 579.83514404
pro = [487.77453613, 508.28781128, 636.34637451, 749.43041992, 877.48901367, 974.54180908]
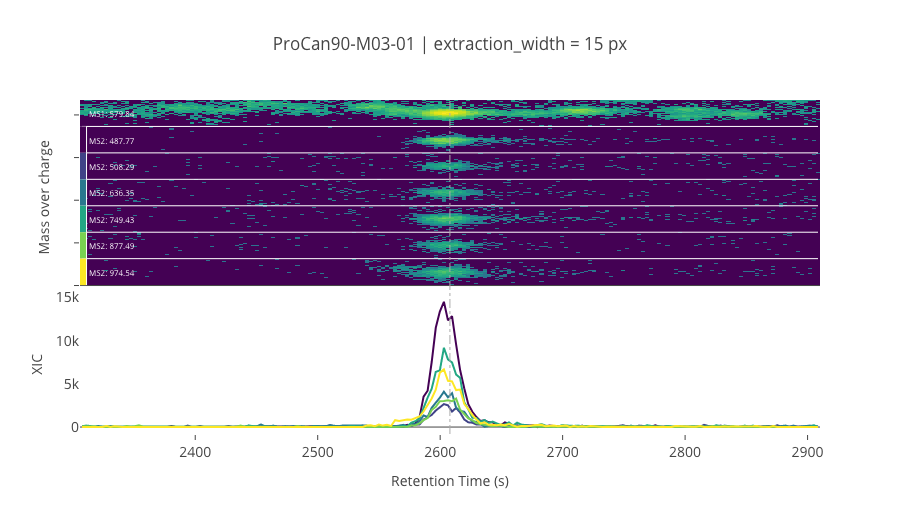
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-048'
peptide_id, peptide, charge, rt, pre = '114292', 'NALESYAFNMK', 2, 2491.33, 644.30554199
pro = [539.26464844, 610.30175781, 773.36505127, 860.39709473, 989.43969727, 1102.5238037]
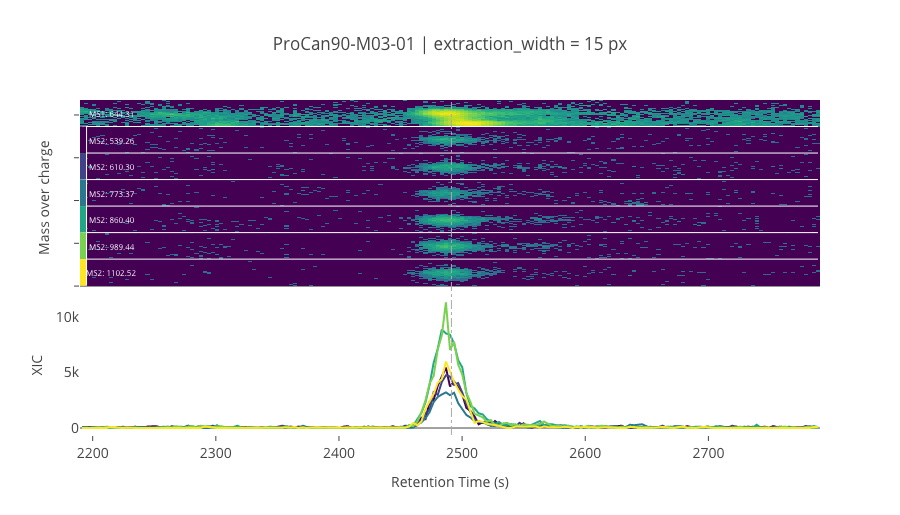
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-049'
peptide_id, peptide, charge, rt, pre = '193918', 'VIHDNFGIVEGLMTTVHAITATQK', 4, 4158.86, 649.59545898
pro = [548.30383301, 579.28851318, 732.42504883, 783.37841797, 869.48394775, 896.46246338]
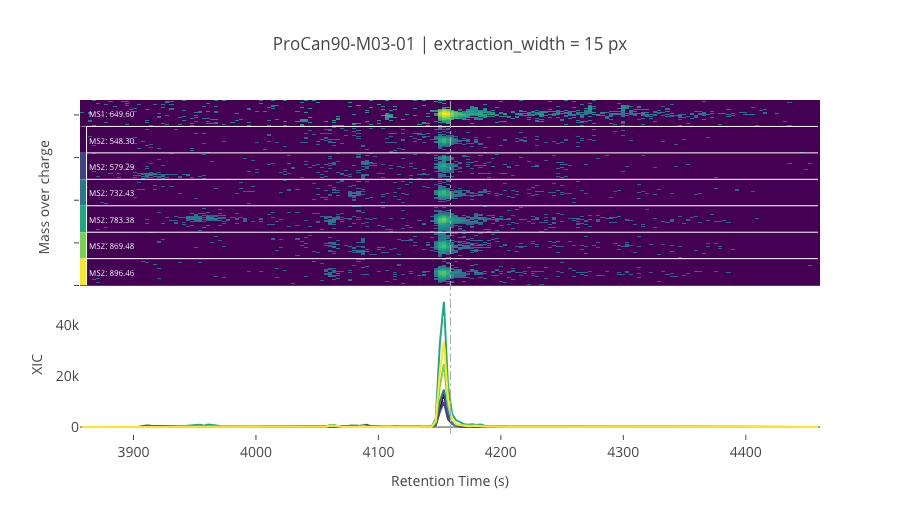
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-055'
peptide_id, peptide, charge, rt, pre = '33530', 'GGGGNFGPGPGSNFR', 2, 1180.7, 689.31835938
pro = [580.28381348, 677.33654785, 734.35803223, 831.4107666, 888.43225098, 1035.5006104]
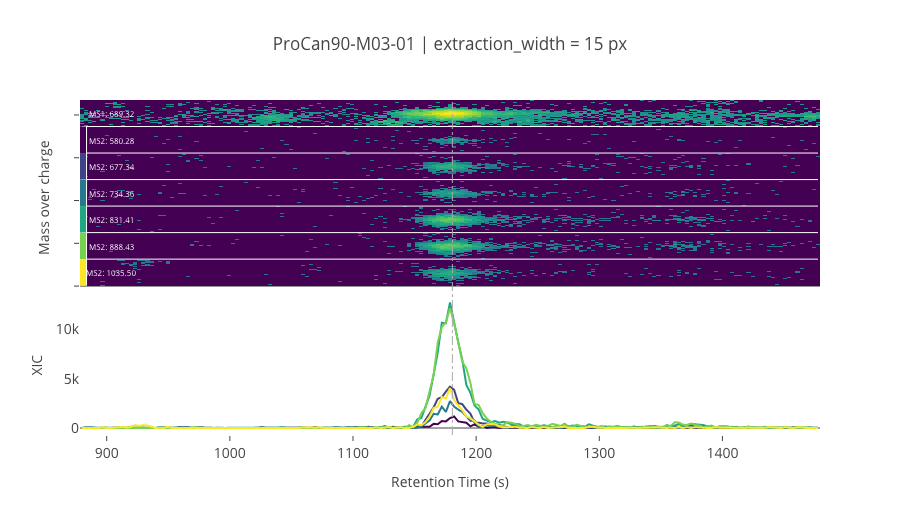
Outliers defined by the C++ library results¶
In fitting an analytic model to the raw data, we now have a new set of parameters that can be used for exploring outliers; where an outlier is defined in a manner similar to plotting the whiskers on a box plot. For certain parameters, if the value from the fit model falls outside a multiplier of the inter-quartile range, then it is classed as an outlier.
[11]:
def plot_params(requantified, cols, colname, whis=5.0):
groups = requantified.groupby('InjectionName')
n_injections = len(groups)
colours = [plt.cm.viridis(i / n_injections) for i in range(n_injections)]
scale = 5
ncols = len(cols)
fig, axes = plt.subplots(ncols=ncols, figsize=(scale * ncols, scale))
for i, (ax, col) in enumerate(zip(axes, cols)):
xlims = []
for colour, (injection, df) in zip(colours, groups):
mp_groups = df[col]
mp_groups.plot(kind='kde', ax=ax, bw_method=0.3, c=colour, label=injection)
# plot outlier limits
q1 = mp_groups.quantile(0.25)
q3 = mp_groups.quantile(0.75)
iqr = q3 - q1
outlier_offset = whis * iqr
lower = q1 - outlier_offset
upper = q3 + outlier_offset
ax.axvline(lower, ls='--', c=colour, alpha=0.5)
ax.axvline(upper, ls='--', c=colour, alpha=0.5)
xlim = list(ax.get_xlim())
xlim[0] = max(xlim[0], lower - 2 * outlier_offset)
xlim[1] = min(xlim[1], upper + 2 * outlier_offset)
xlims.append(xlim)
ax.set_xlim((min(x[0] for x in xlims), max(x[1] for x in xlims)))
ax.set_title(col)
if i == 0:
ax.legend()
plt.show()
def mark_outliers(requantified, col, whis=5.0):
mp_groups = requantified[col].groupby(requantified.InjectionName)
mean = mp_groups.mean()
# calculate outliers as you would in a box plot
q1 = mp_groups.quantile(0.25)
q3 = mp_groups.quantile(0.75)
iqr = q3 - q1
lower = q1 - whis * iqr
upper = q3 + whis * iqr
delta_col = 'Delta' + col
requantified[delta_col] = pd.np.NaN
outlier_col = 'Outlier' + col
requantified[outlier_col] = pd.np.NaN
for n in lower.index:
mask = requantified.InjectionName == n
requantified.loc[mask, delta_col] = (requantified.loc[mask, col] - mean.loc[n]).abs()
requantified.loc[mask, outlier_col] = (
(requantified.loc[mask, col] < lower.loc[n]) |
(requantified.loc[mask, col] > upper.loc[n])
)
def get_outlier_peptides(requantified, injection_name, n=8, force=False, **kwargs):
col_mz0 = 'ModelParamMz0MS2'
col_rt0 = 'ModelParamRT0'
col_sigma_rt = 'ModelParamSigmaRT'
for col in [col_mz0, col_rt0, col_sigma_rt]:
delta_col = 'Delta' + col
outlier_col = 'Outlier' + col
if delta_col not in requantified.columns or force:
mark_outliers(requantified, col, **kwargs)
outlier_col = 'PotentialOutlier'
requantified[outlier_col] = (
requantified['Outlier' + col_mz0] |
requantified['Outlier' + col_rt0] |
requantified['Outlier' + col_sigma_rt]
)
display(requantified[outlier_col].groupby([
requantified['InjectionName'],
requantified[outlier_col],
]).count())
delta_col = 'PotentialOutlierDelta'
requantified[delta_col] = (
requantified['Delta' + col_mz0] *
requantified['Delta' + col_rt0] *
requantified['Delta' + col_sigma_rt]
)
outlier_peptide_ids = requantified.loc[
(requantified.InjectionName == injection_name) &
requantified[outlier_col]
].sort_values([delta_col, 'Intensity'], ascending=(False, False)).head(n)
return outlier_peptide_ids.peptide_id.tolist()
SGS data¶
[12]:
plot_params(
sgs_requantified,
['ModelParamSigmaRT', 'ModelParamSigmaMz', 'ModelParamRT0', 'ModelParamMz0MS1', 'ModelParamMz0MS2'],
'ModelParam',
)
sgs_outlier_peptide_ids = get_outlier_peptides(
requantified=sgs_requantified,
injection_name=f'{sgs_file:03d}',
force=True,
whis=5.0,
)
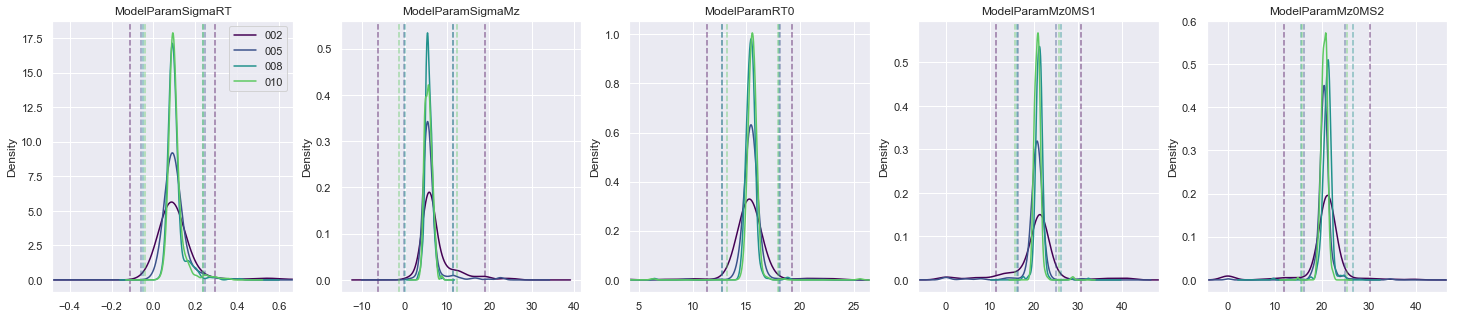
InjectionName PotentialOutlier
002 False 161
True 22
005 False 258
True 18
008 False 331
True 8
010 False 331
True 12
Name: PotentialOutlier, dtype: int64
[13]:
explore_raw_data(
file=sgs_file,
quantifiers=sgs_quantifiers,
peptide_ids=sgs_outlier_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-011'
peptide_id, peptide, charge, rt, pre = '1158', 'YYDYTLSINGK(UniMod:259)', 2, 3657.93, 672.832
pro = [526.307, 639.392, 740.439, 1018.53]
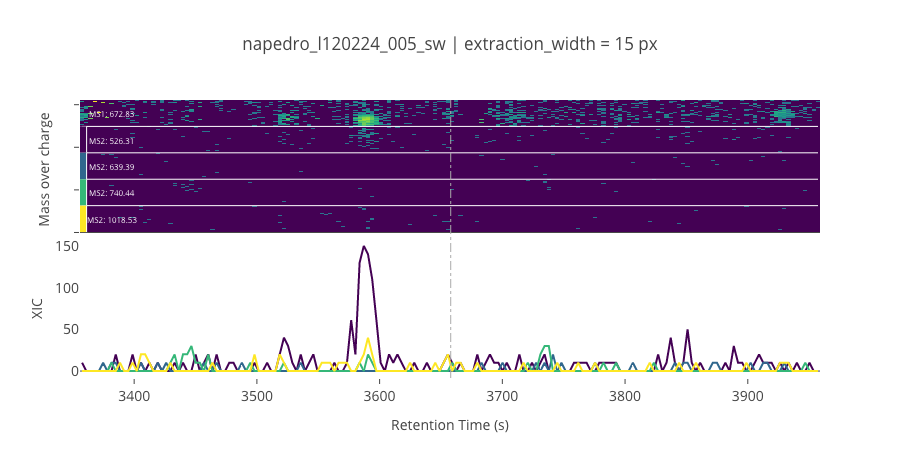
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-012'
peptide_id, peptide, charge, rt, pre = '985', 'TIVGVSEDFAALR(UniMod:267)', 2, 4574.05, 694.376
pro = [587.354, 918.455, 1074.55, 1173.61]
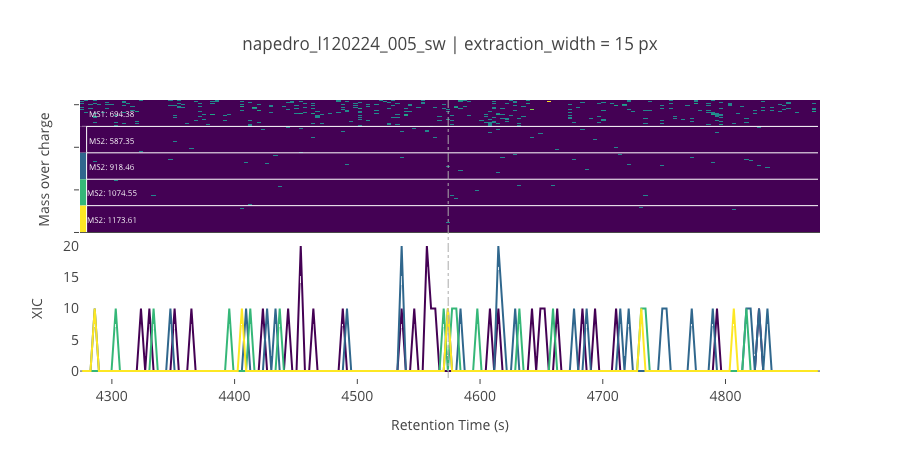
--------------------------------------------------
ms2_name = 'ms2-012'
peptide_id, peptide, charge, rt, pre = '1051', 'VESPVLPPVLVPK(UniMod:259)', 2, 4738.54, 691.431
pro = [757.506, 870.59, 1066.71, 1153.74]
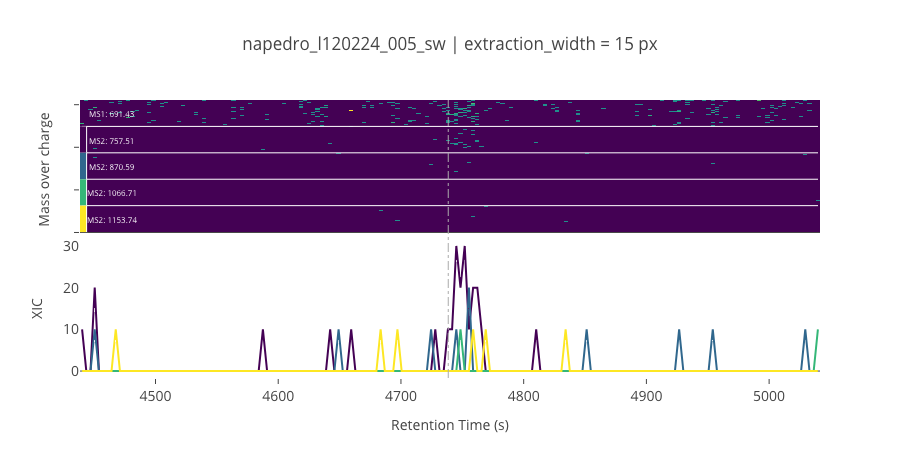
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-014'
peptide_id, peptide, charge, rt, pre = '212', 'EYTEGVNGQPSIR(UniMod:267)', 2, 2438.35, 730.356
pro = [667.376, 781.419, 937.509, 1066.55]
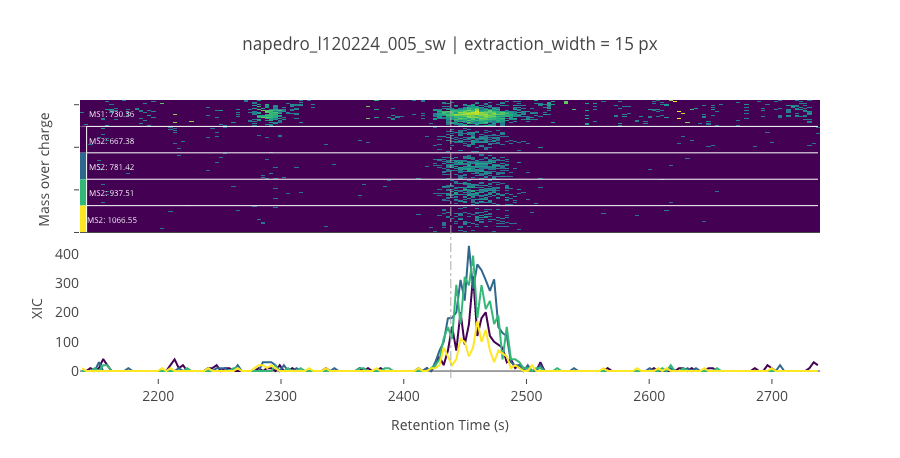
--------------------------------------------------
ms2_name = 'ms2-014'
peptide_id, peptide, charge, rt, pre = '695', 'MIVDPVEPHGEMK(UniMod:259)', 2, 2851.66, 745.367
pro = [706.343, 835.386, 1031.51, 1245.6]
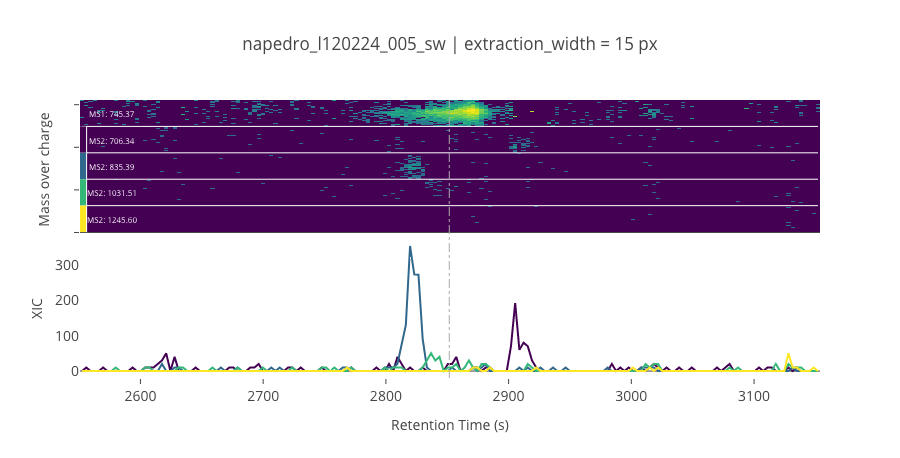
--------------------------------------------------
ms2_name = 'ms2-014'
peptide_id, peptide, charge, rt, pre = '920', 'SDSSDTPPLPSPPGK(UniMod:259)', 2, 2804.66, 745.367
pro = [800.476, 897.528, 998.576, 1113.6]
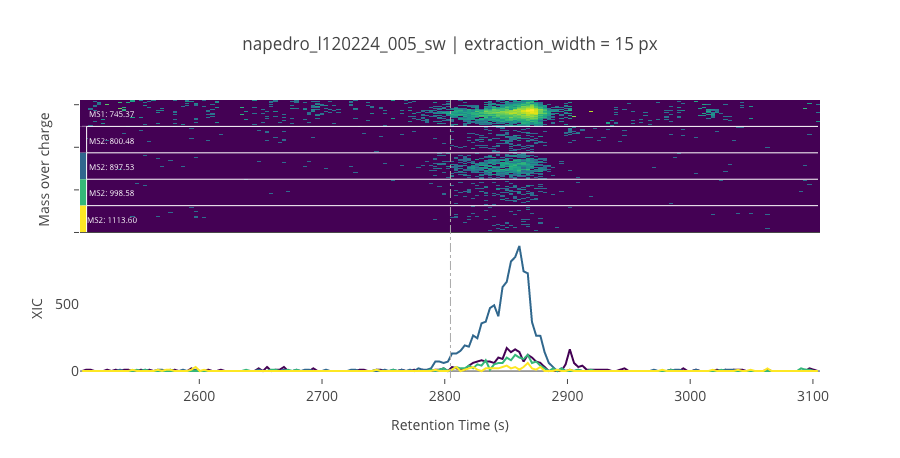
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-015'
peptide_id, peptide, charge, rt, pre = '239', 'FPSPVSHADDLYGK(UniMod:259)', 2, 3293.18, 770.88
pro = [789.387, 1013.48, 1209.6, 1296.63]
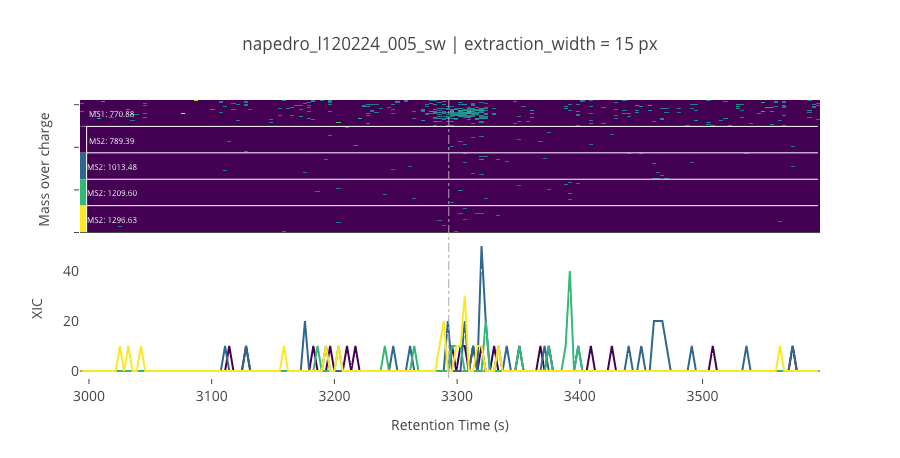
--------------------------------------------------
ms2_name = 'ms2-015'
peptide_id, peptide, charge, rt, pre = '729', 'NTDFNGVNNIHQK(UniMod:259)', 2, 2128.47, 754.87
pro = [647.371, 917.504, 1031.55, 1178.62]
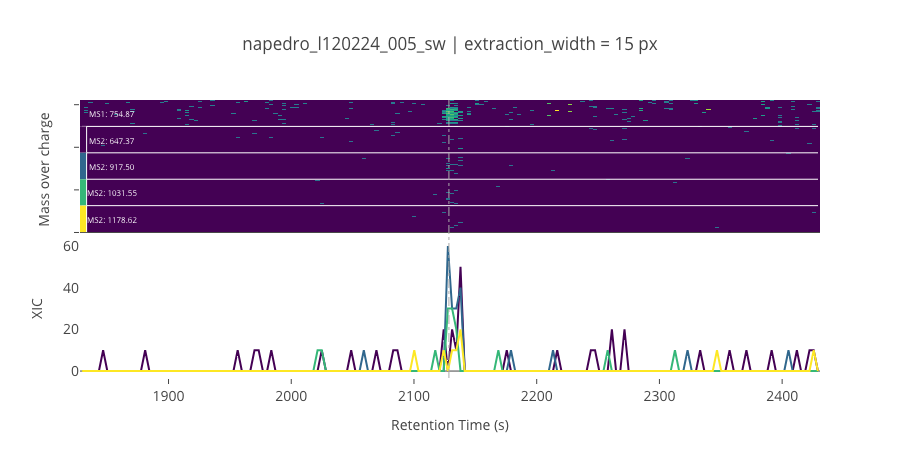
ProCan90 data¶
[14]:
plot_params(
procan90_requantified,
['ModelParamSigmaRT', 'ModelParamSigmaMz', 'ModelParamRT0', 'ModelParamMz0MS1', 'ModelParamMz0MS2'],
'ModelParam',
)
procan90_outlier_peptide_ids = get_outlier_peptides(
requantified=procan90_requantified,
injection_name=f'ProCan90-M{procan90_file}',
force=True,
)
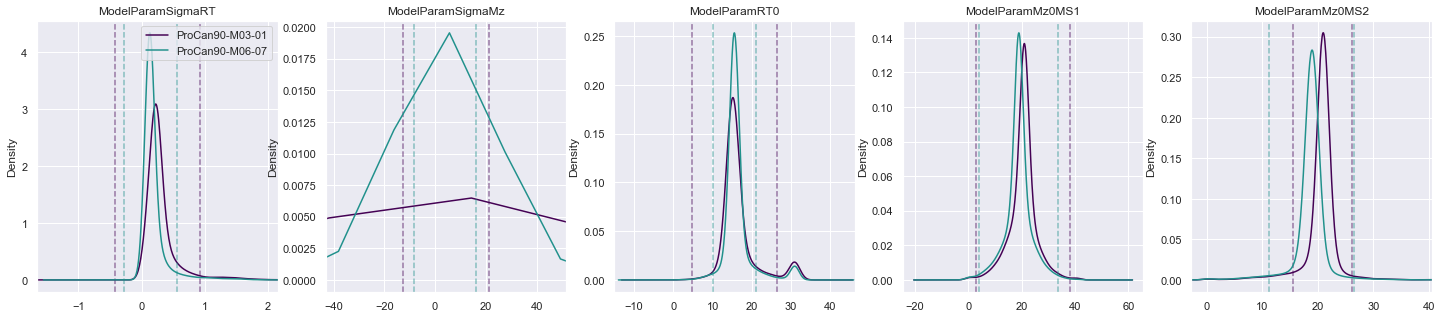
InjectionName PotentialOutlier
ProCan90-M03-01 False 17022
True 2377
ProCan90-M06-07 False 23297
True 3138
Name: PotentialOutlier, dtype: int64
[15]:
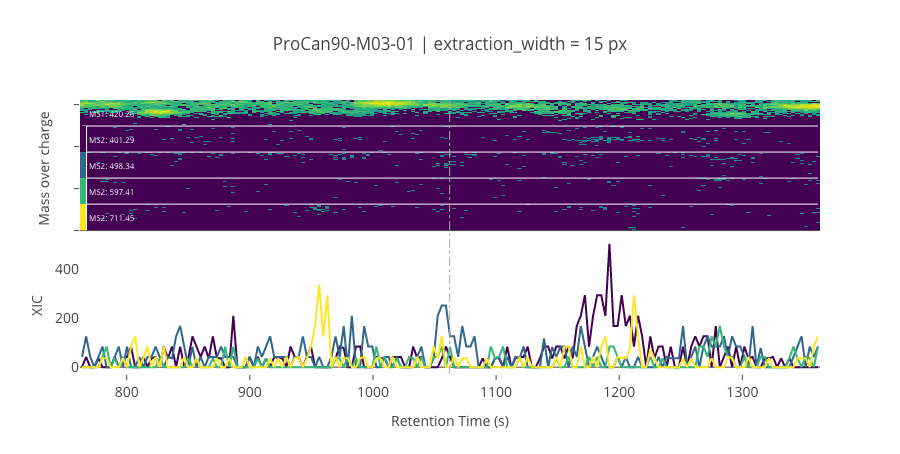
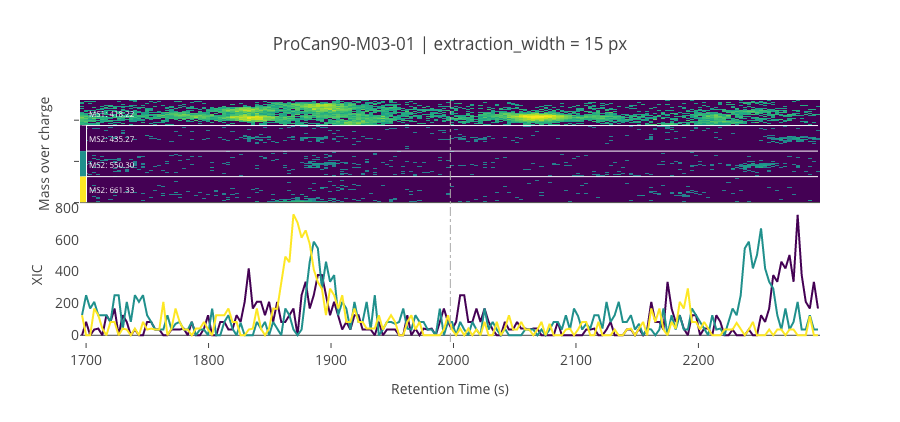
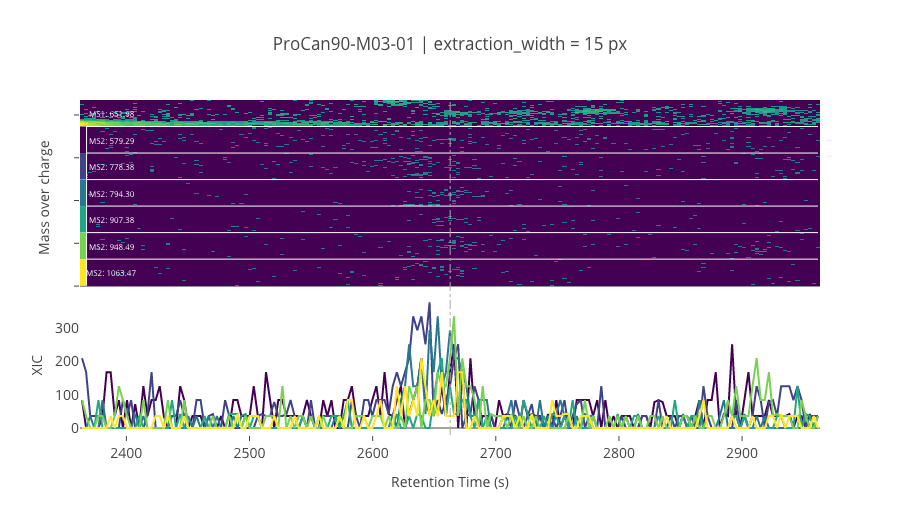
explore_raw_data(
file=procan90_file,
quantifiers=procan90_quantifiers,
peptide_ids=procan90_outlier_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-004'
peptide_id, peptide, charge, rt, pre = '122878', 'QNVPIIR', 2, 1062.29, 420.2585144
pro = [401.28707886, 498.33984375, 597.40826416, 711.45117188]
--------------------------------------------------
ms2_name = 'ms2-004'
peptide_id, peptide, charge, rt, pre = '132921', 'REDFLR', 2, 1997.44, 418.22467041
pro = [435.27142334, 550.29840088, 661.3303833]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-049'
peptide_id, peptide, charge, rt, pre = '38727', 'HGEPEEDIVGLQAFQER', 3, 2662.94, 651.98156738
pro = [579.28851318, 778.38421631, 794.29516602, 907.37921143, 948.48974609, 1063.4691162]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-050'
peptide_id, peptide, charge, rt, pre = '114165', 'NADMQEELIASLEEQLK', 3, 4136.85, 654.3225708
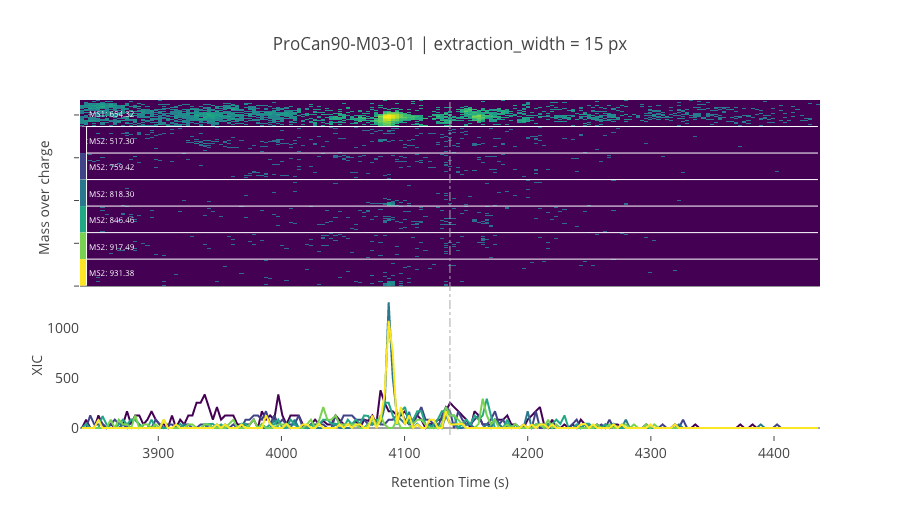
pro = [517.29803467, 759.42468262, 818.29852295, 846.45672607, 917.49383545, 931.38256836]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-057'
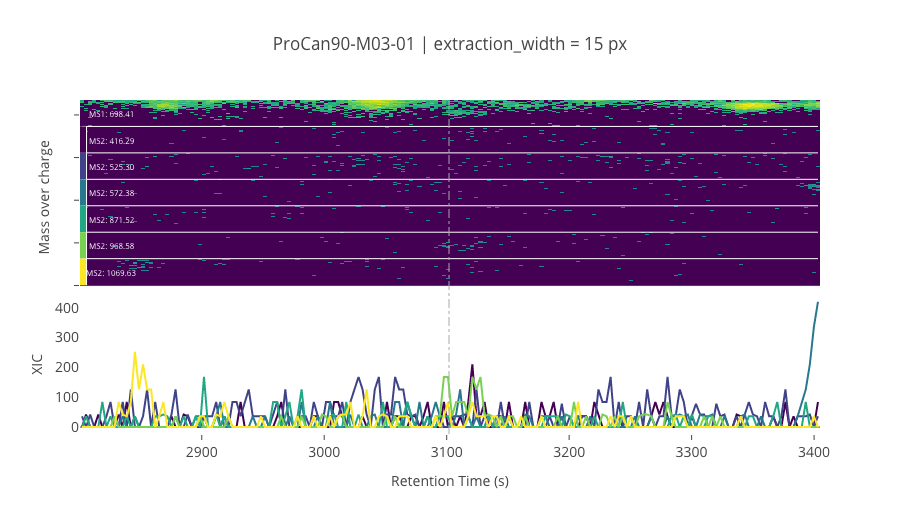
peptide_id, peptide, charge, rt, pre = '195660', 'VNITPAEVGVLVGK', 2, 3101.9, 698.41394043
pro = [416.28674316, 525.30310059, 572.37664795, 871.52471924, 968.57751465, 1069.6252441]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-064'
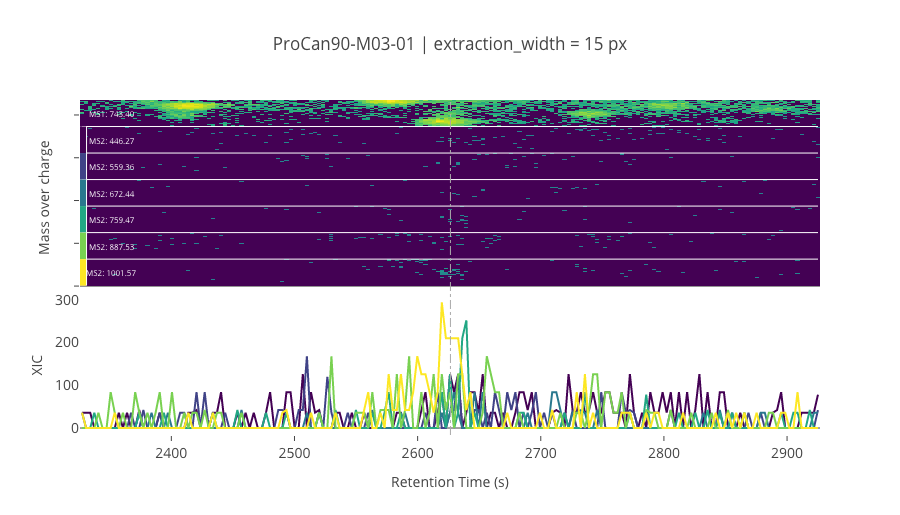
peptide_id, peptide, charge, rt, pre = '18270', 'EAGNINQSLLTLGR', 2, 2626.65, 743.40460205
pro = [446.27215576, 559.35620117, 672.44030762, 759.47229004, 887.53088379, 1001.5737915]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-089'
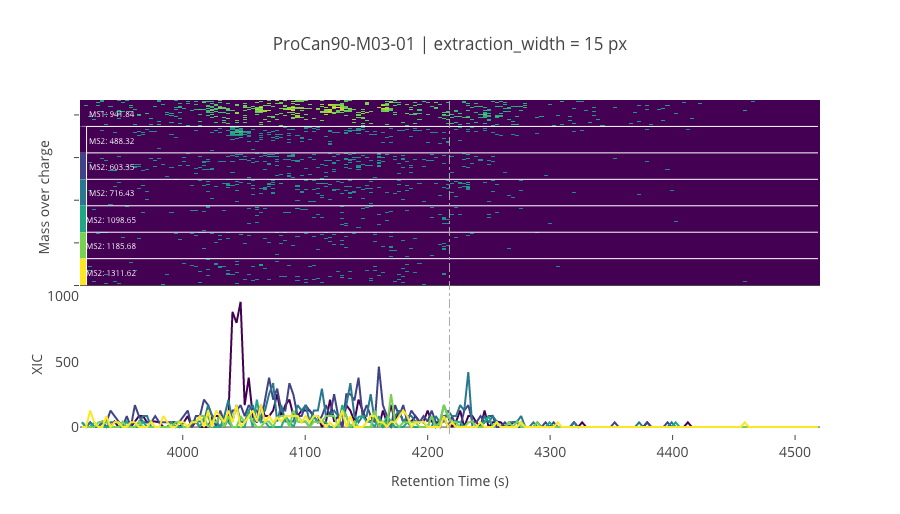
peptide_id, peptide, charge, rt, pre = '192141', 'VEEASPGRPSSVDTLLSPTALIDSILR', 3, 4217.8, 941.84143066
pro = [488.31912231, 603.34606934, 716.43011475, 1098.6517334, 1185.6837158, 1311.6175537]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-096'
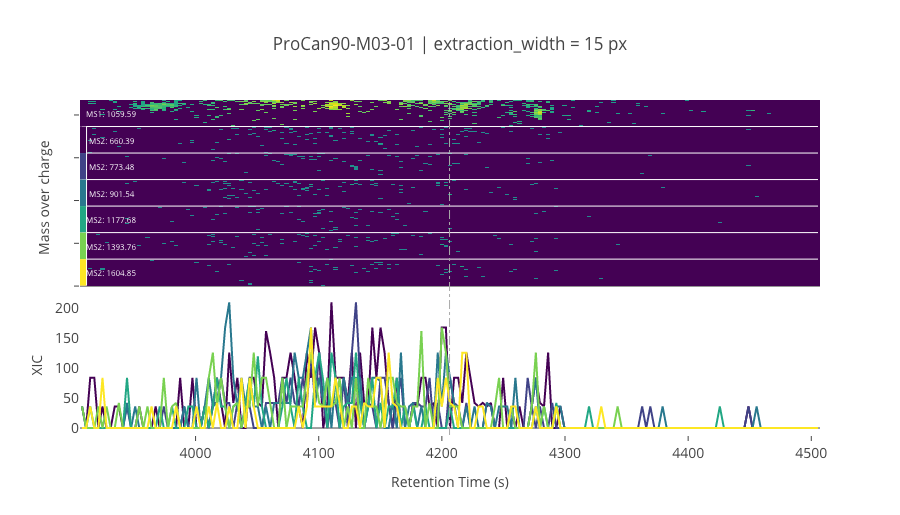
peptide_id, peptide, charge, rt, pre = '179998', 'SLLSIPNTDYIQLLSEIAK', 2, 4206.23, 1059.5882568
pro = [660.39263916, 773.47674561, 901.53533936, 1177.6827393, 1393.7573242, 1604.8530273]
Building a python model¶
[16]:
import scipy
import scipy.optimize
import scipy.special
class Requant():
def __init__(
self,
tof_fname,
rt_pixel_half_width=15,
mz_pixel_half_width=20,
num_ms1_isotopes=1,
debug_plots=False,
):
self.swath_run = toffee.SwathRun(tof_fname)
self.ms1_swath_map = self.swath_run.loadSwathMap(toffee.ToffeeWriter.MS1_NAME)
self.rt_pixel_half_width = rt_pixel_half_width
self.mz_pixel_half_width = mz_pixel_half_width
self.num_ms1_isotopes = num_ms1_isotopes
self.debug_plots = debug_plots
def run(self, ms2_name, peak_groups):
ms2_swath_map = self.swath_run.loadSwathMap(ms2_name)
results = []
for peak_group in peak_groups:
assert isinstance(peak_group, requant.RequantPeakGroup)
raw_data = self._extract_raw_data(ms2_swath_map, peak_group)
if raw_data is None:
continue
ms1_intensity, ms2_intensity = self._fit_intensities(*raw_data)
results.append((peak_group.modifiedSequence(), peak_group.charge(), ms1_intensity, ms2_intensity))
return results
def _extract_raw_data(self, ms2_swath_map, peak_group):
rt_range = toffee.RetentionTimeRangeWithPixelHalfWidth(
peak_group.retentionTime(),
self.rt_pixel_half_width,
)
expected_shape = (2 * self.rt_pixel_half_width + 1, 2 * self.mz_pixel_half_width + 1)
raw_data = []
normalisers = []
delta_rt = []
delta_mz = []
def extract(sm, mz):
mz_range = toffee.MassOverChargeRangeWithPixelHalfWidth(mz, self.mz_pixel_half_width)
chrom = sm.filteredExtractedIonChromatogram(mz_range, rt_range)
if chrom.intensities.shape == expected_shape:
r = chrom.intensities.astype(np.float)
n = max(1, r.max())
raw_data.append(r / n)
normalisers.append(n)
delta_rt.append(chrom.retentionTime[1] - chrom.retentionTime[0])
delta_mz.append(np.diff(chrom.massOverCharge).mean())
else:
print(mz, chrom.intensities.shape, '!=', expected_shape)
# MS1 data
for i in range(self.num_ms1_isotopes + 1):
isotope_mass_offset = 1.0033548
mz = peak_group.massOverCharge() + i * isotope_mass_offset / peak_group.charge()
extract(self.ms1_swath_map, mz)
if len(raw_data) == 0:
return None
# MS2 data
for fragment_mz in peak_group.fragmentMassOverCharges():
extract(ms2_swath_map, fragment_mz)
return delta_rt, delta_mz, raw_data, normalisers
def _fit_intensities(self, delta_rt, delta_mz, raw_data, normalisers):
n_ms1 = self.num_ms1_isotopes + 1
n_frag = len(raw_data)
n_rt = self.rt_pixel_half_width * 2 + 1
n_mz = self.mz_pixel_half_width * 2 + 1
assert n_mz > n_rt
n_slice = n_rt * n_mz
rt = np.arange(n_rt) + 0.5
mz = np.arange(n_mz) + 0.5
rt, mz = np.meshgrid(rt, mz, indexing='ij')
raw_data_vec = np.zeros((n_frag * n_rt * n_mz,))
rt_vec = np.zeros_like(raw_data_vec)
mz_vec = np.zeros_like(raw_data_vec)
for i_frag, raw in enumerate(raw_data):
idx = slice(i_frag * n_slice, (i_frag + 1) * n_slice)
raw_data_vec[idx] = raw.ravel()
rt_vec[idx] = rt.ravel()
mz_vec[idx] = mz.ravel()
for i_rt in range(n_rt):
for i_mz in range(n_mz):
idx = i_frag * n_slice + i_rt * n_mz + i_mz
assert raw_data_vec[idx] == raw[i_rt, i_mz]
assert rt_vec[idx] == rt[i_rt, i_mz]
assert mz_vec[idx] == mz[i_rt, i_mz]
# pre-allocate for fast calculations
fit_data_vec = np.zeros_like(raw_data_vec)
sigma_rt_vec = np.zeros_like(raw_data_vec)
sigma_mz_vec = np.zeros_like(raw_data_vec)
rt0_vec = np.zeros_like(raw_data_vec)
mz0_vec = np.zeros_like(raw_data_vec)
amplitude_vec = np.zeros_like(raw_data_vec)
chem_noise_amplitude_vec = np.zeros_like(raw_data_vec)
radius_vec = np.zeros_like(raw_data_vec)
def sigmoid(n, x):
# n * scipy.special.expit(x)
return 0.5 * n * (1 + np.tanh(x))
def guassian_2d(sigma_rt, sigma_mz, rt0, mz0_ms1, mz0_ms2, amplitude, chem_noise_amplitude):
# set up vectors, enforce positivity and ranges
rt0_vec[:] = sigmoid(n_rt, rt0)
sigma_rt_vec[:] = sigma_rt ** 2
sigma_mz_vec[:] = sigma_mz ** 2
for i_frag in range(n_frag):
idx = slice(i_frag * n_slice, (i_frag + 1) * n_slice)
amplitude_vec[idx] = amplitude[i_frag] ** 2
if i_frag < n_ms1:
mz0_vec[idx] = sigmoid(n_mz, mz0_ms1)
chem_noise_amplitude_vec[idx] = chem_noise_amplitude[i_frag] ** 2
else:
mz0_vec[idx] = sigmoid(n_mz, mz0_ms2)
# calculate the fit model
exp_rt = -0.5 * ((np.log(rt_vec) - np.log(rt0_vec)) / sigma_rt_vec) ** 2
denom_rt = np.sqrt(2 * np.pi) * rt_vec * sigma_rt_vec
exp_mz = -0.5 * ((mz_vec - mz0_vec) / sigma_mz_vec) ** 2
signal_vec = amplitude_vec * np.exp(exp_rt + exp_mz) / denom_rt
chem_noise_vec = chem_noise_amplitude_vec * np.exp(exp_mz)
return signal_vec + chem_noise_vec
def all_guassian_2d(sigma_rt, sigma_mz, rt0, mz0_ms1, mz0_ms2, *amplitudes_and_noise):
assert len(amplitudes_and_noise) == n_frag + n_ms1
fit_data_vec[:] = guassian_2d(
sigma_rt,
sigma_mz,
rt0,
mz0_ms1,
mz0_ms2,
amplitudes_and_noise[:n_frag],
amplitudes_and_noise[n_frag:],
)
def residual(params):
all_guassian_2d(*params)
return fit_data_vec - raw_data_vec
# sigma_x, sigma_y, x0, y0_ms1, y0_ms2
guess = [0.5, 2.0, 0.0, 0.0, 0.0]
n = len(guess)
# add amplitude vector
guess = guess + [1.0] * n_frag
# add chemical noise vector
guess = guess + [0.1] * n_ms1
result = scipy.optimize.least_squares(residual, guess, method='lm')
ms1_intensity, ms2_intensity = np.NaN, np.NaN
if result.success:
# print(result)
# print('')
# print('n_frag', n_frag)
# print('sigma_x, sigma_y, x0', result.x[:n])
# print('y0', result.x[n:n + n_frag])
# print('amplitude', result.x[n + n_frag:n + 2 * n_frag])
# print('noise', result.x[n + 2 * n_frag:])
if self.debug_plots:
self._make_debug_plots(raw_data_vec, fit_data_vec)
# calculate final fit
params = result.x.copy()
params[-n_ms1:] = 0.0 # set checmical noise to zero
all_guassian_2d(*params)
area_under_curve = []
raw_area_under_curve = []
for i_frag in range(n_frag):
idx = slice(i_frag * n_slice, (i_frag + 1) * n_slice)
a = fit_data_vec[idx].sum()
a *= normalisers[i_frag]
a *= delta_rt[i_frag]
a *= delta_mz[i_frag]
area_under_curve.append(a)
a = raw_data_vec[idx].sum()
a *= normalisers[i_frag]
a *= delta_rt[i_frag]
a *= delta_mz[i_frag]
raw_area_under_curve.append(a)
# integrate intensity -- subtracting noise
ms1_intensity = sum(area_under_curve[:n_ms1])
ms2_intensity = sum(area_under_curve[n_ms1:])
raw_ms1_intensity = sum(raw_area_under_curve[:n_ms1])
raw_ms2_intensity = sum(raw_area_under_curve[n_ms1:])
print('Raw MS1 & MS2:', raw_ms1_intensity, raw_ms2_intensity)
print('Fit MS1 & MS2:', ms1_intensity, ms2_intensity)
return ms1_intensity, ms2_intensity
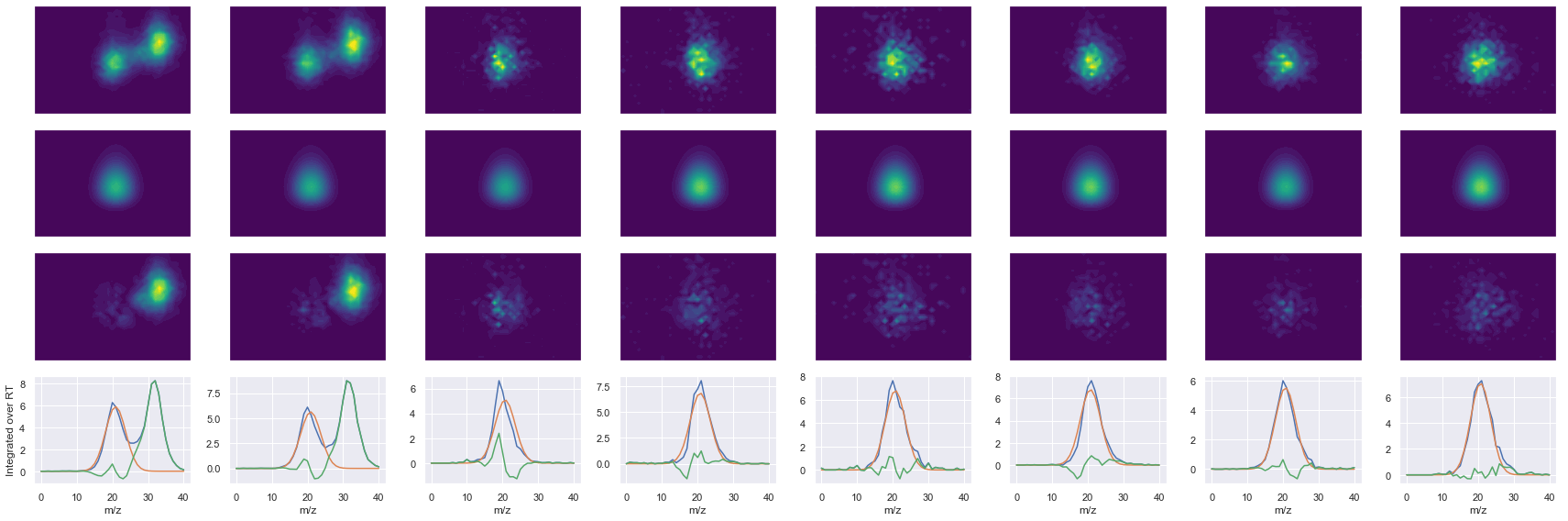
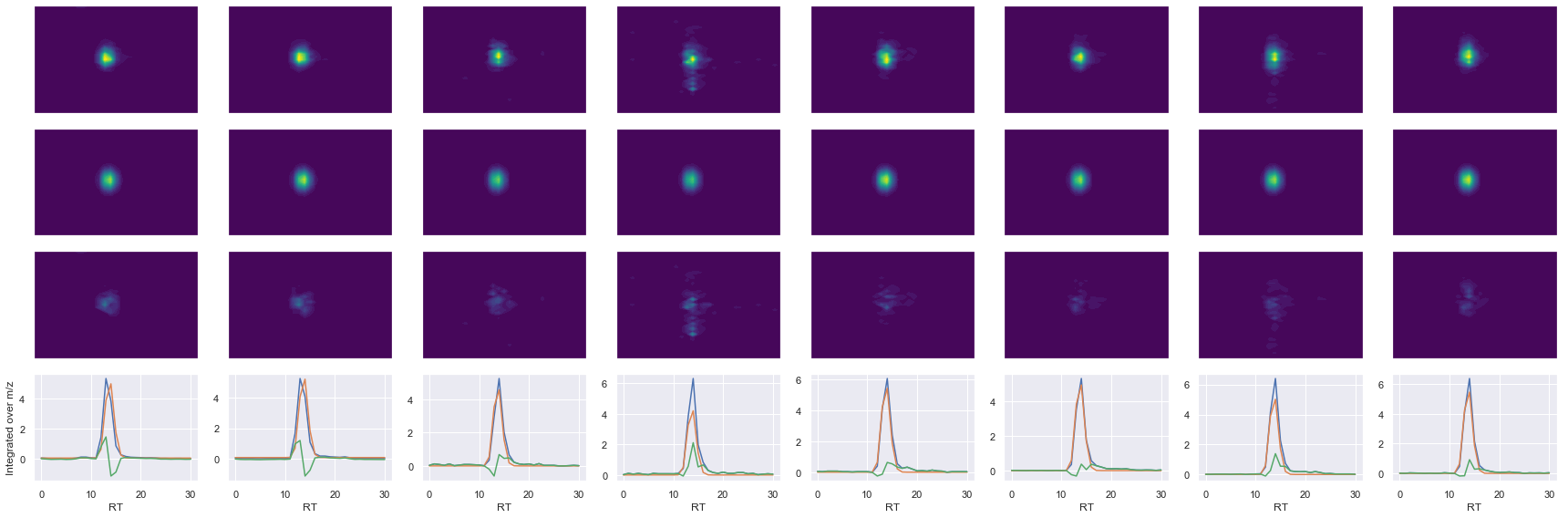
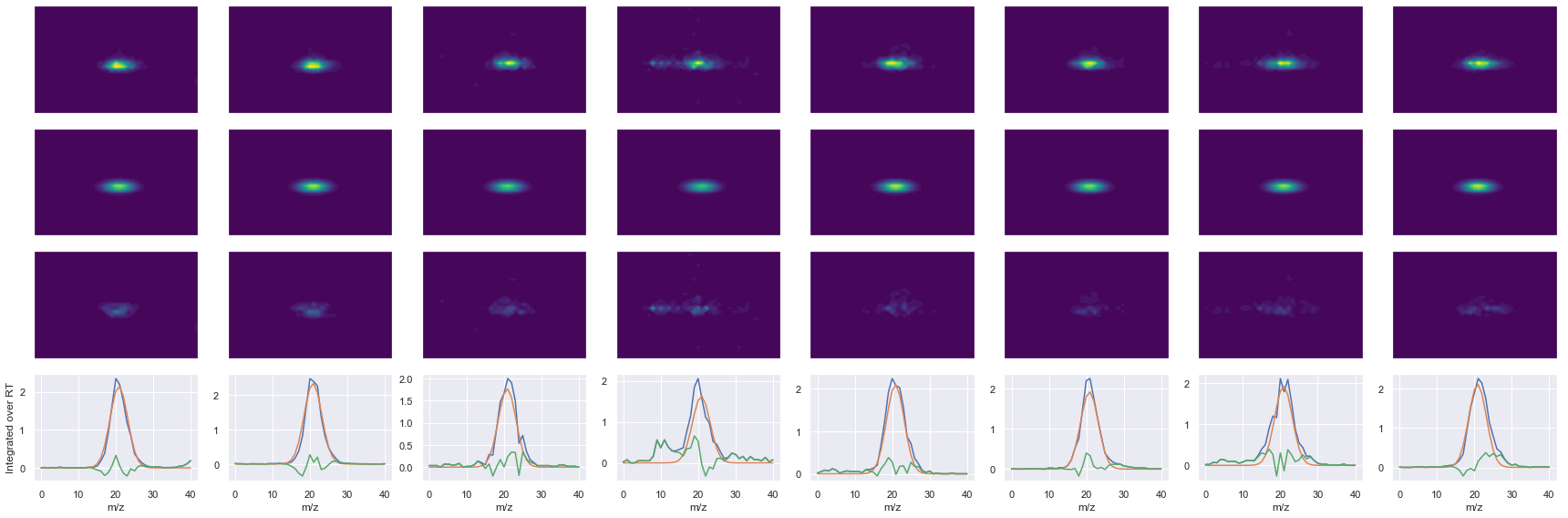
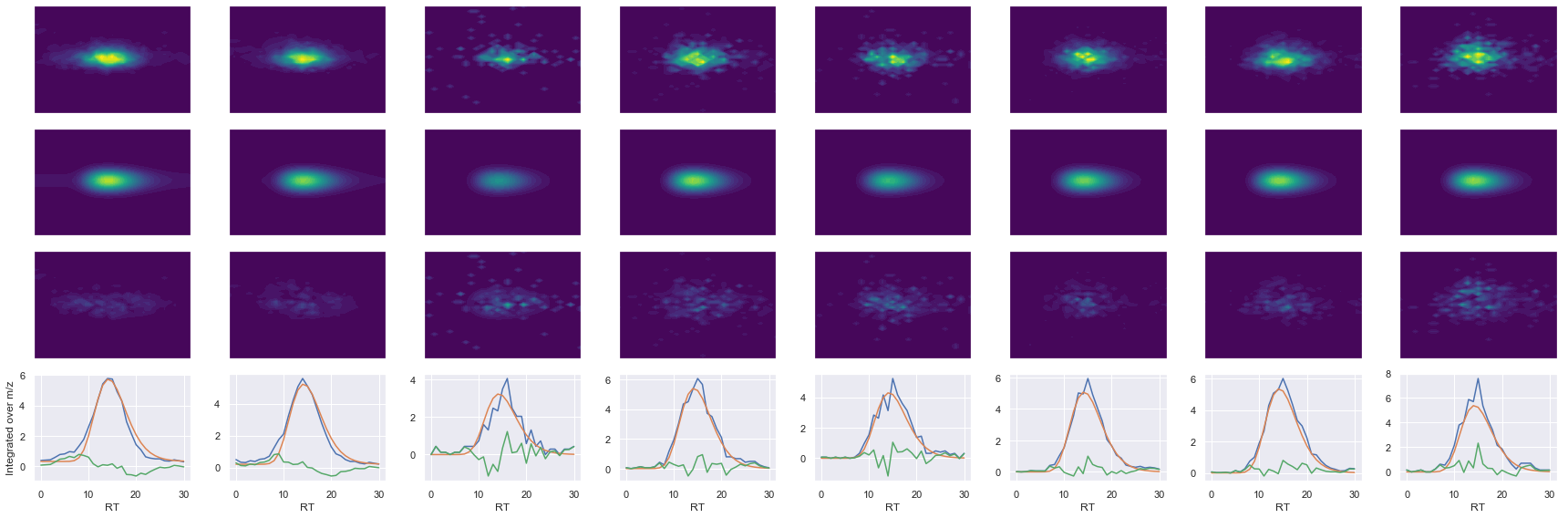
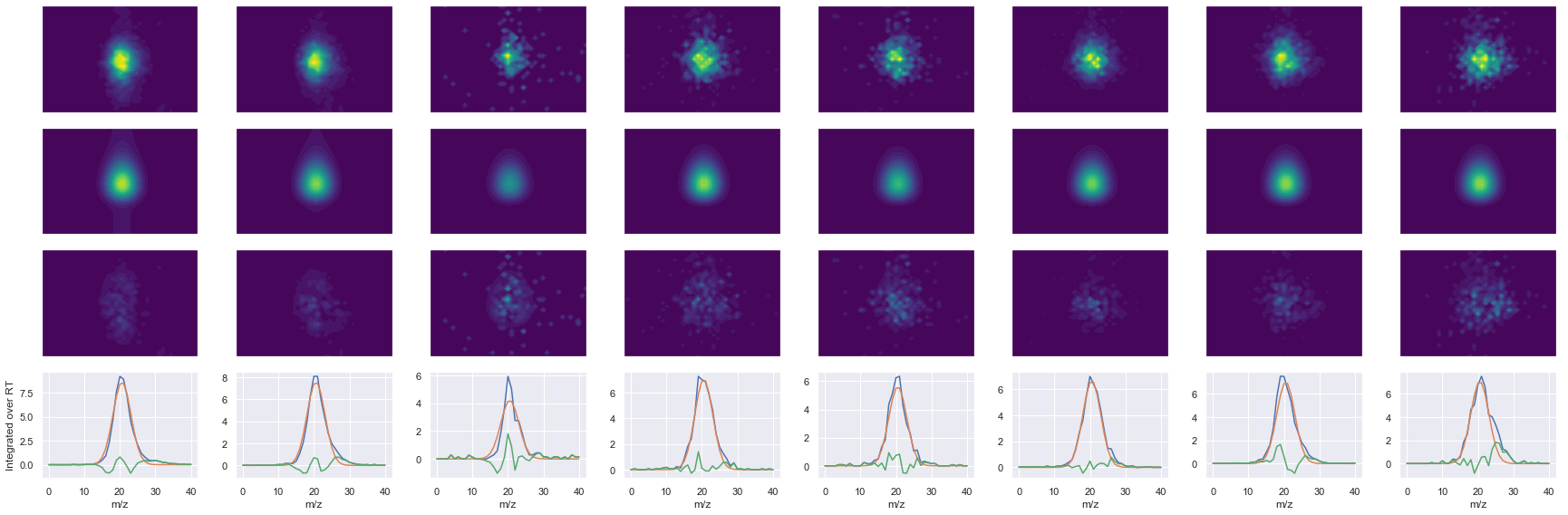
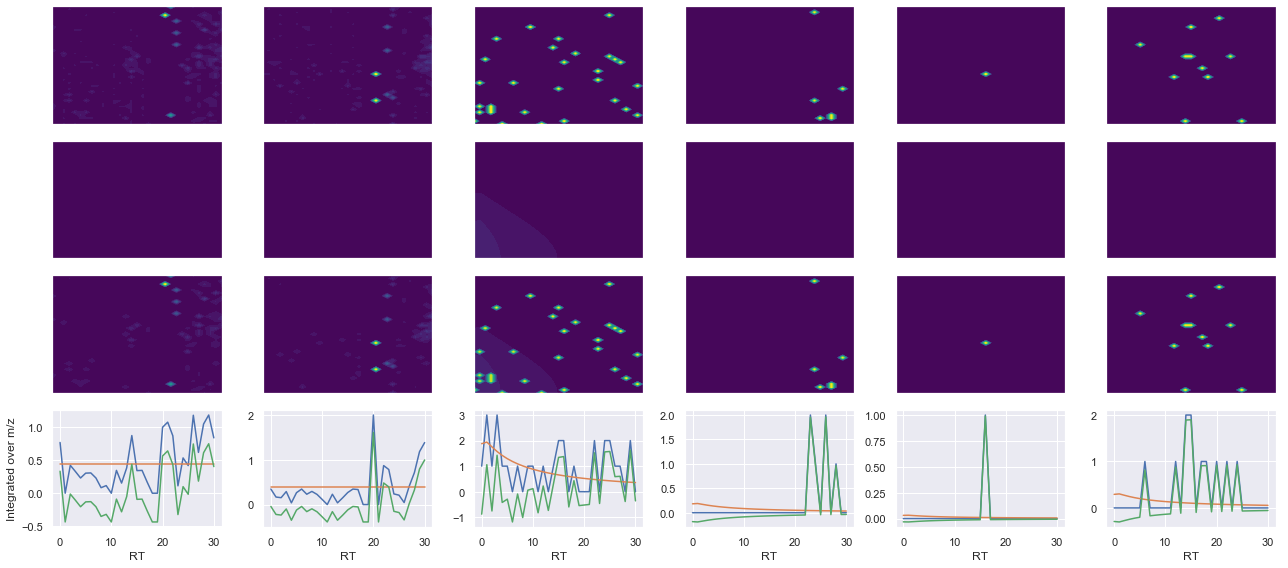
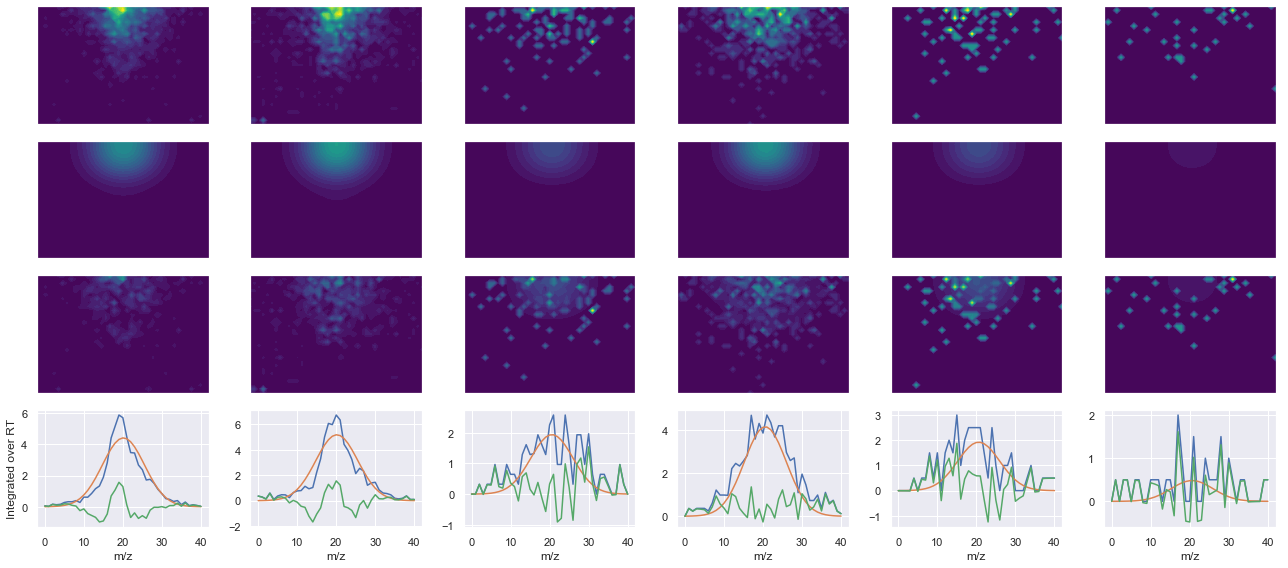
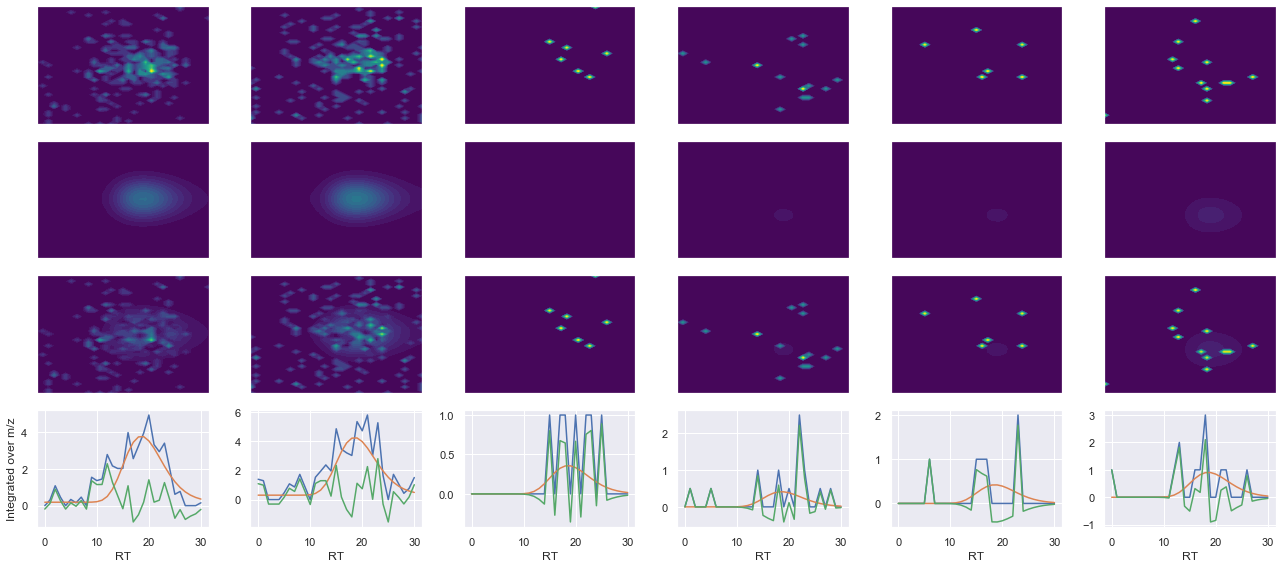
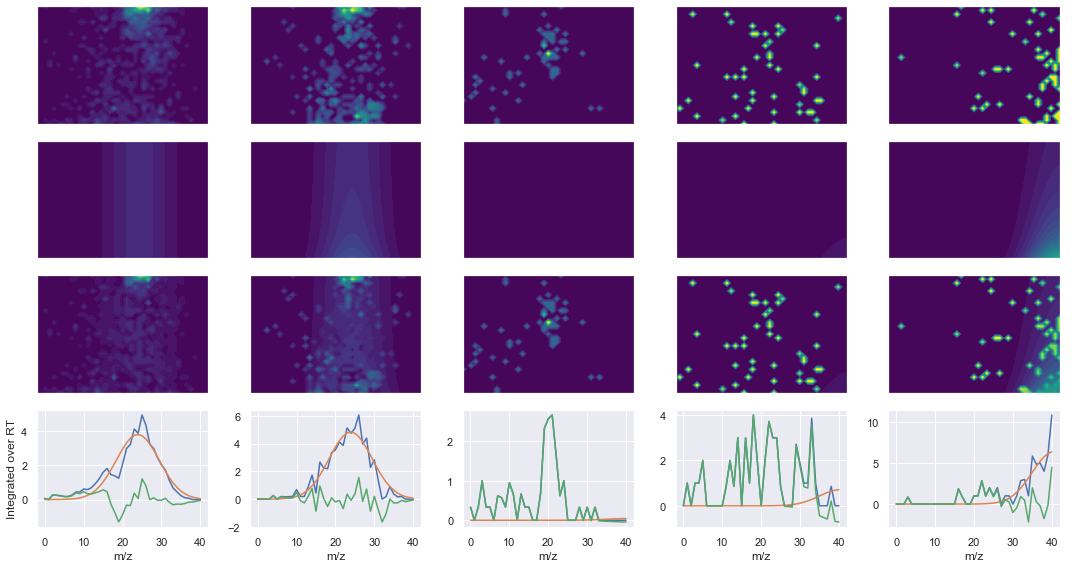
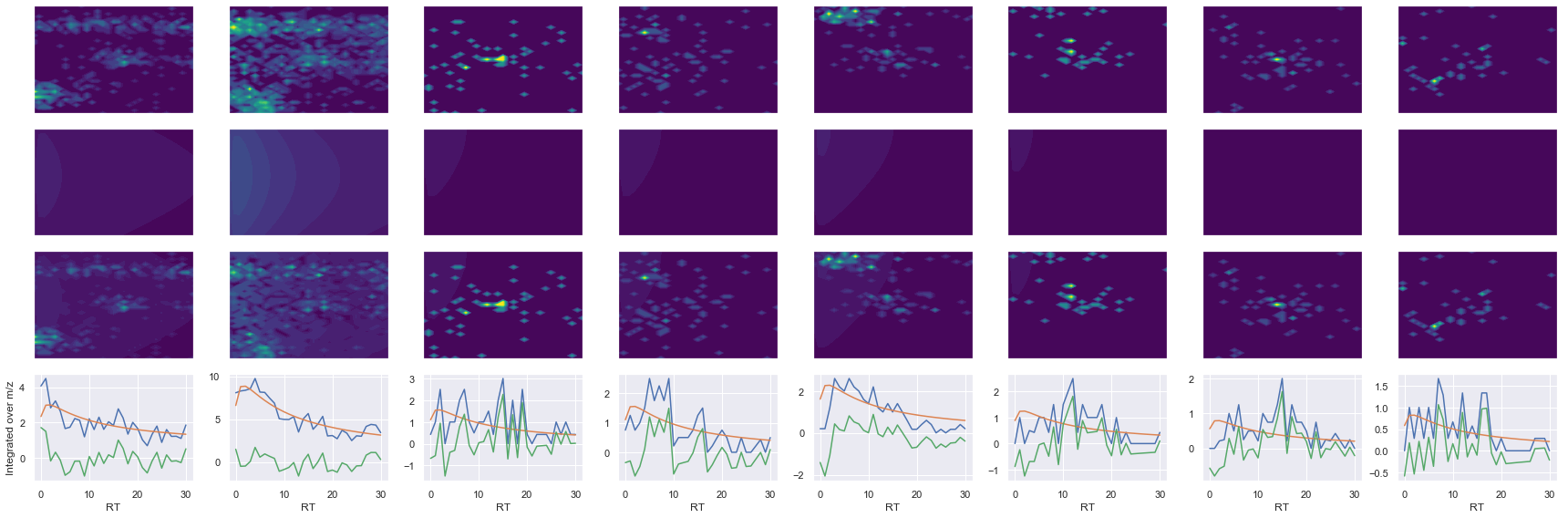
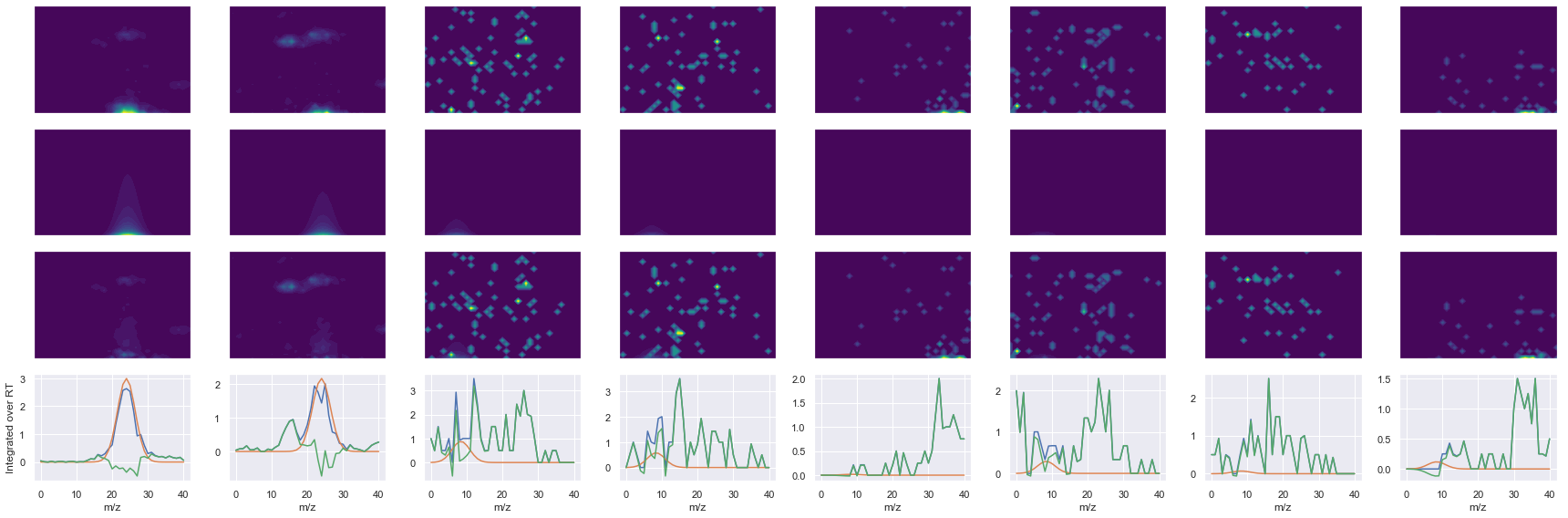
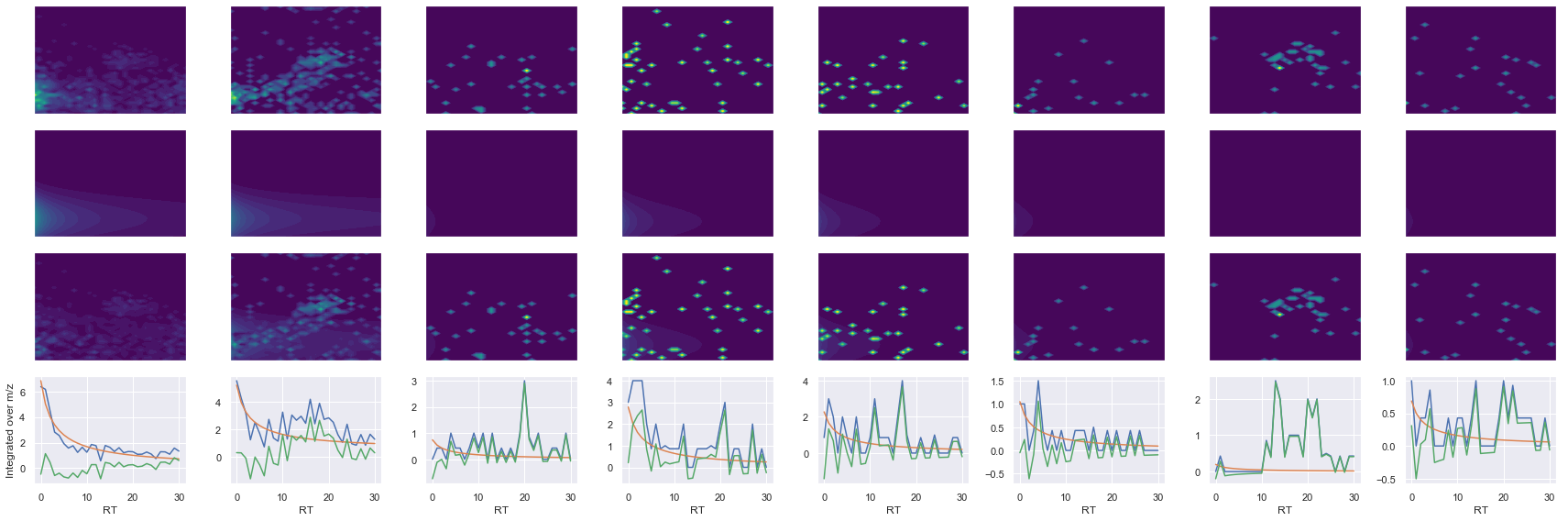
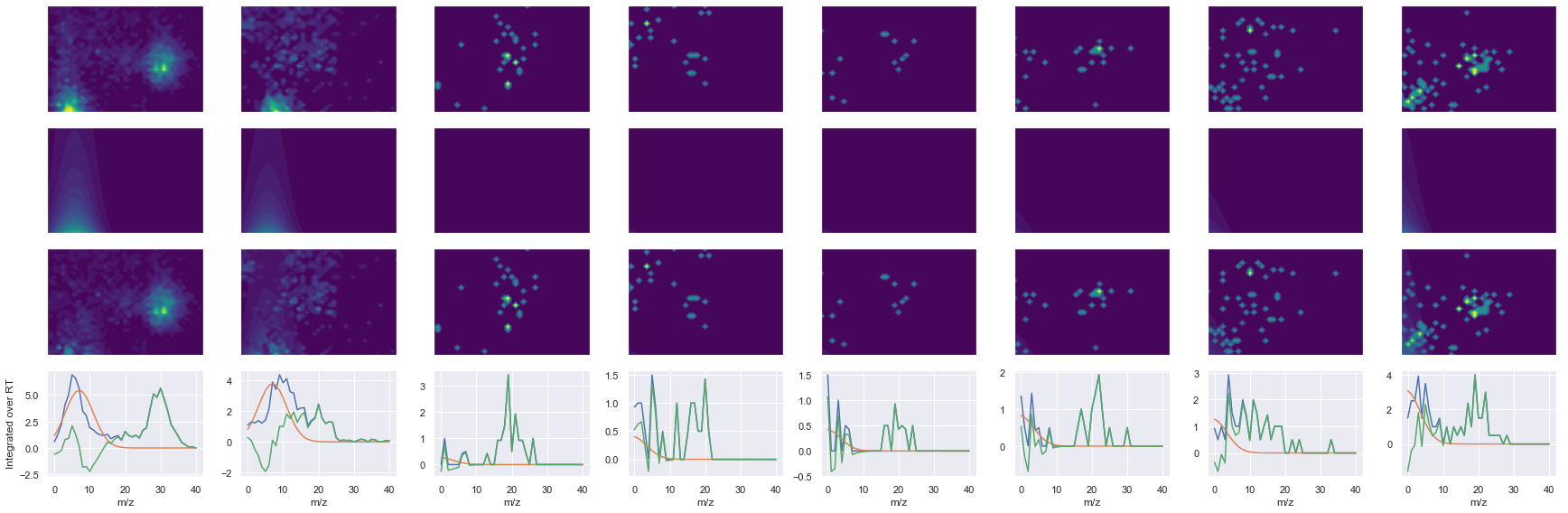
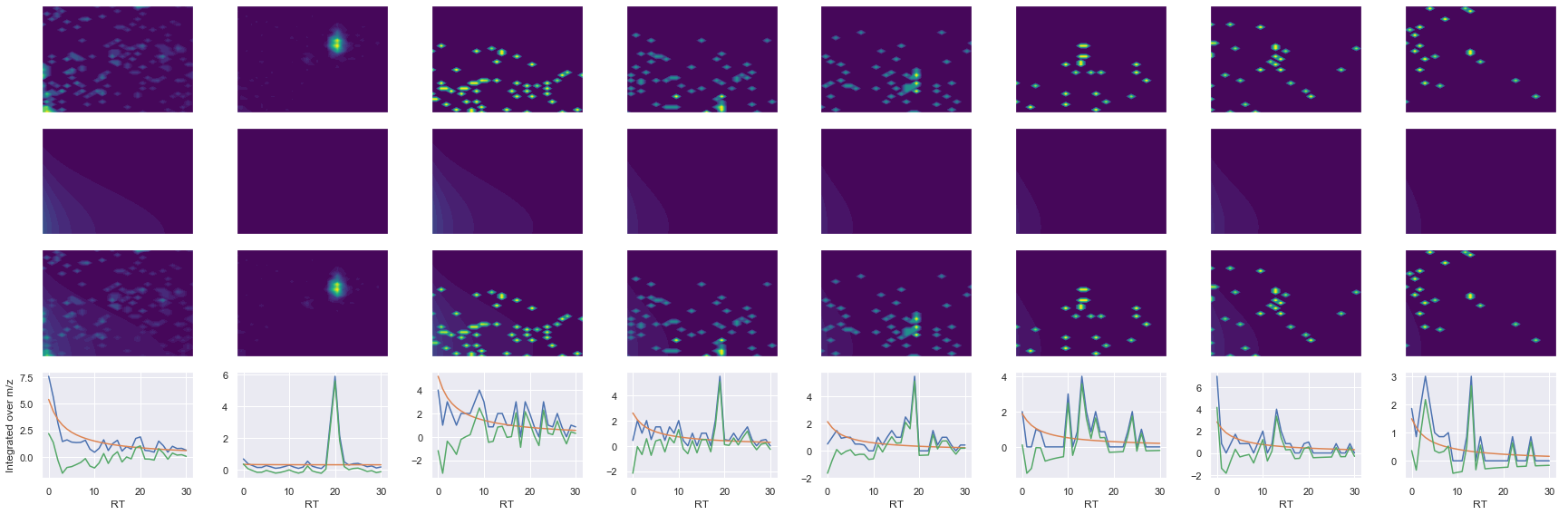
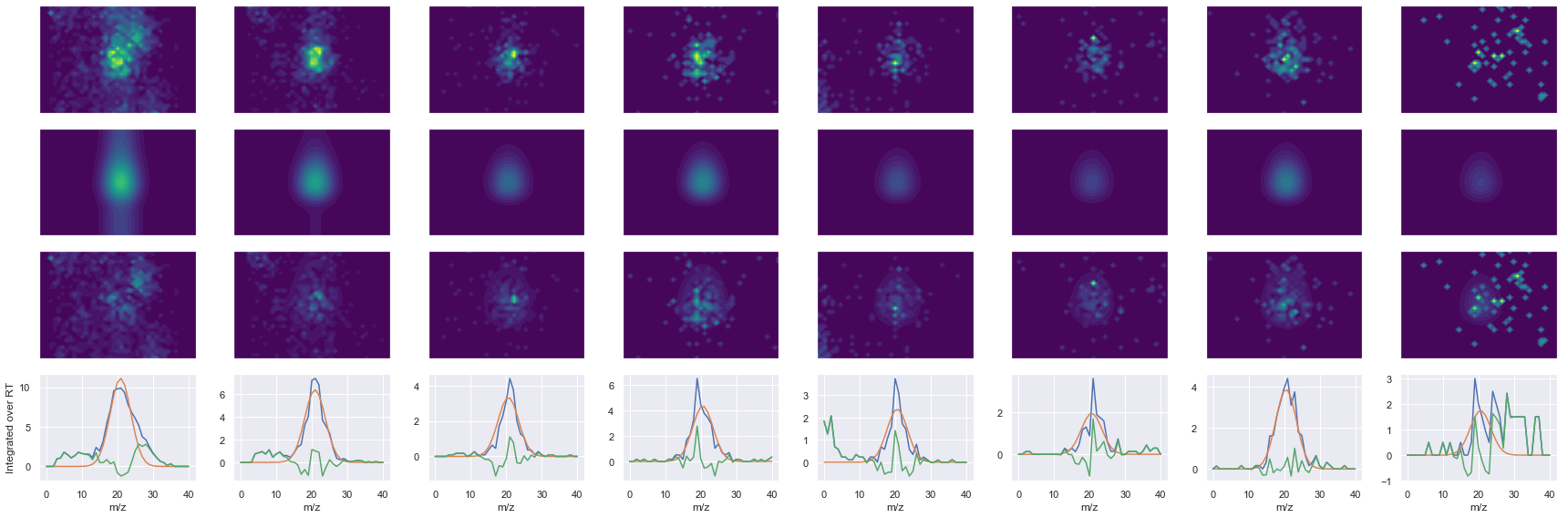
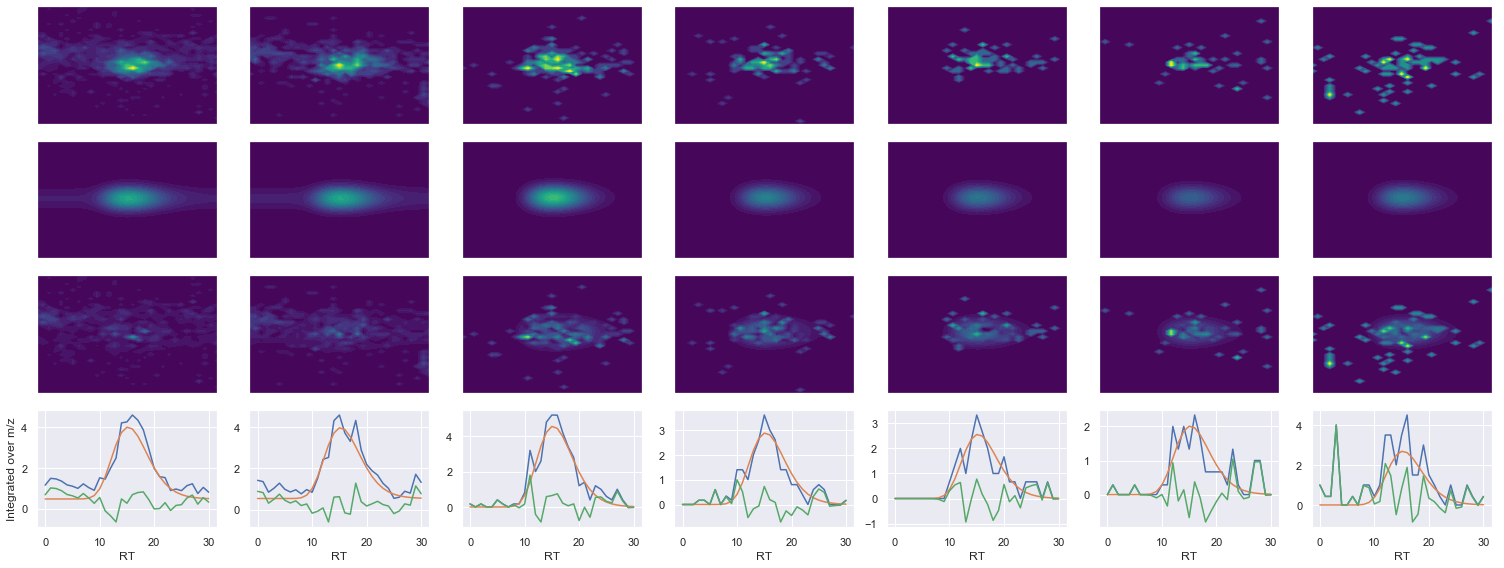
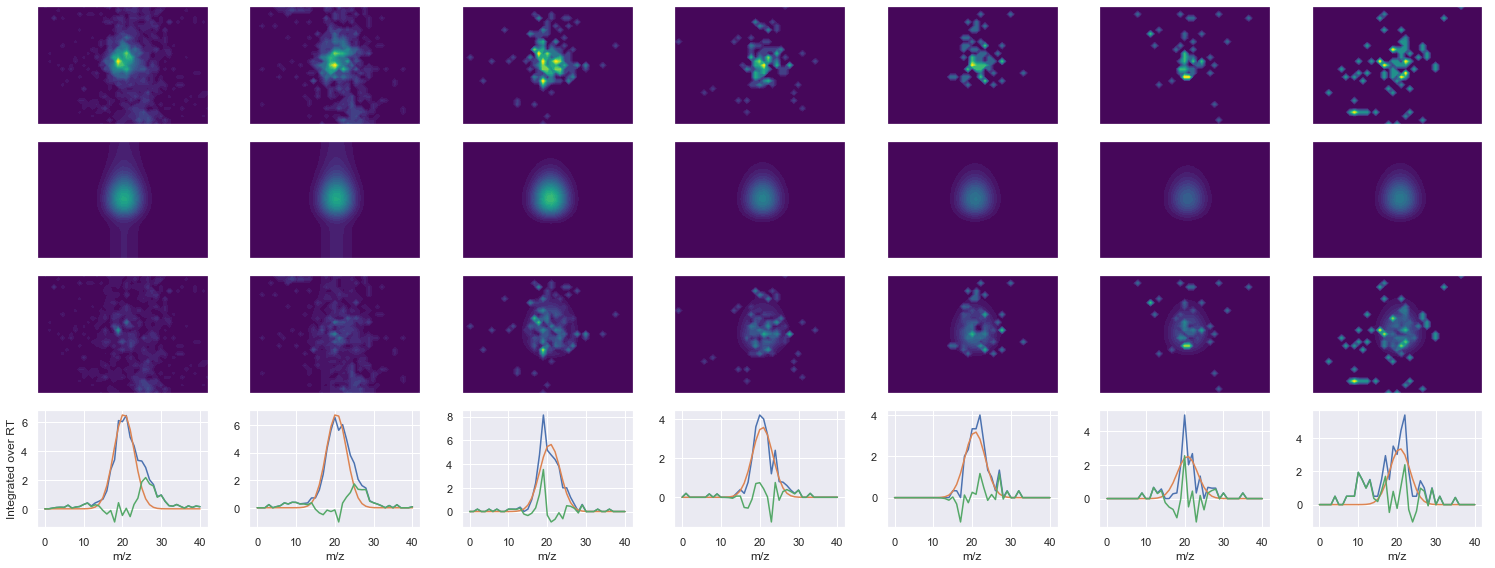
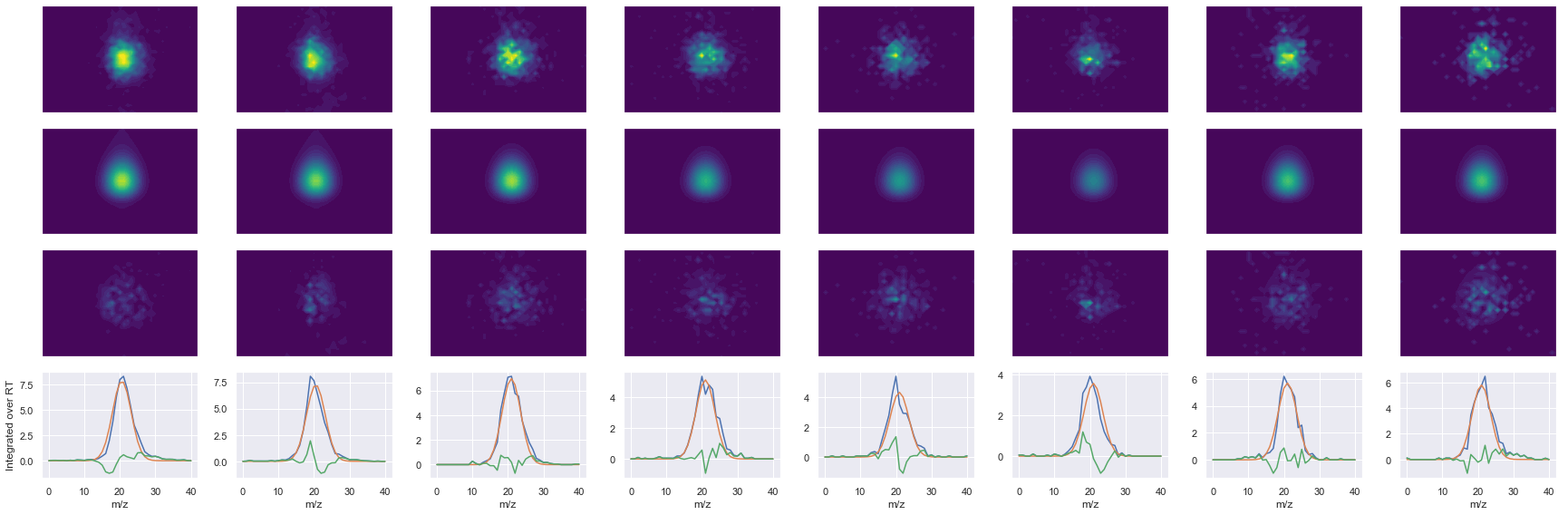
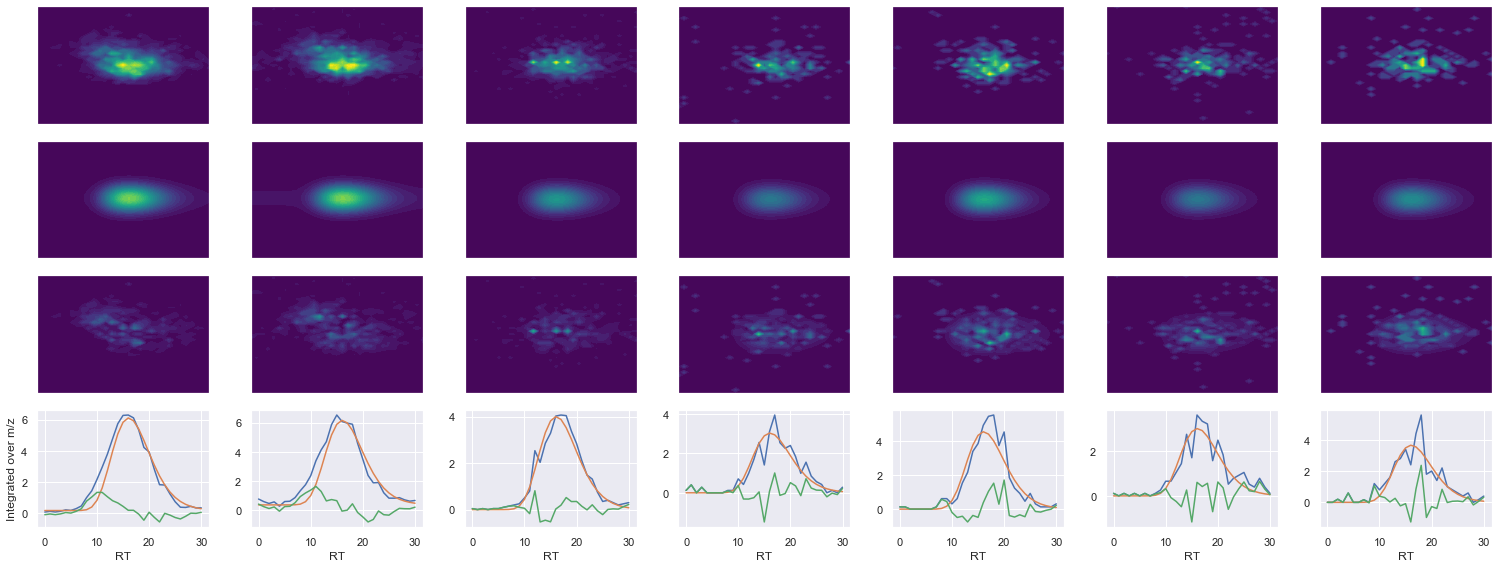
def _make_debug_plots(self, raw_data_vec, fit_data_vec):
import matplotlib.pyplot as plt
n_rt = self.rt_pixel_half_width * 2 + 1
n_mz = self.mz_pixel_half_width * 2 + 1
assert n_mz > n_rt
n_slice = n_rt * n_mz
n_frag = raw_data_vec.shape[0] // n_slice
raw_data = []
fit_data = []
for i_frag in range(n_frag):
idx = slice(i_frag * n_slice, (i_frag + 1) * n_slice)
raw_data.append(raw_data_vec[idx].reshape(n_rt, n_mz))
fit_data.append(fit_data_vec[idx].reshape(n_rt, n_mz))
ncols = len(raw_data)
nrows = 4
scale = 2
figsize = (scale * ncols * 1.5, scale * nrows)
cmap = plt.cm.viridis
for take_transpose in [False, True]:
fig, axes = plt.subplots(nrows=nrows, ncols=ncols, figsize=figsize)
for raw, fit, (ax1, ax2, ax3, ax4) in zip(raw_data, fit_data, axes.T):
x = np.arange(n_rt)
y = np.arange(n_mz)
if take_transpose:
raw = raw.T
fit = fit.T
x, y = y, x
diff = raw - fit
vmin, vmax = 0, max(raw.max(), fit.max())
levels = np.linspace(vmin, vmax, num=30)
ax1.contourf(x, y, raw.T, vmin=vmin, vmax=vmax, levels=levels, cmap=cmap)
ax2.contourf(x, y, fit.T, vmin=vmin, vmax=vmax, levels=levels, cmap=cmap)
ax3.contourf(x, y, np.abs(diff).T, vmin=vmin, vmax=vmax, levels=levels, cmap=cmap)
for ax in (ax1, ax2, ax3):
ax.get_xaxis().set_visible(False)
ax.get_yaxis().set_visible(False)
ax4.plot(x, np.sum(raw, axis=1))
ax4.plot(x, np.sum(fit, axis=1))
ax4.plot(x, np.sum(diff, axis=1))
x_label = 'RT' if not take_transpose else 'm/z'
y_label = 'RT' if take_transpose else 'm/z'
axes[0, 0].set_ylabel(f'Raw data ({y_label})')
axes[1, 0].set_ylabel(f'Fit data ({y_label})')
axes[2, 0].set_ylabel(f'Difference ({y_label})')
axes[3, 0].set_ylabel(f'Integrated over {y_label}')
for ax in axes[-1, :]:
ax.set_xlabel(x_label)
fig.tight_layout()
plt.show()
def run_python_requant(file, quantifiers, peptide_ids=None):
q = quantifiers[file]
swath_run = toffee.SwathRun(q.toffee_filename)
srl = q._add_ms2_windows(swath_run, q.srl)
if peptide_ids is None:
peptide_ids = q.scores \
.sort_values(['m_score', 'Intensity'], ascending=(True, False)) \
.iloc[100:115] \
.peptide_id \
.tolist()
all_srl = srl.loc[srl.peptide_id.isin(peptide_ids)].copy()
operator = Requant(q.toffee_filename, debug_plots=True)
for ms2_name in sorted(all_srl.MS2Name.unique()):
print('*' * 80)
ms2_srl = all_srl.loc[all_srl.MS2Name == ms2_name]
for peptide_id in ms2_srl.peptide_id.unique():
print('-' * 50)
fragments = ms2_srl.loc[ms2_srl.peptide_id == peptide_id]
pre = fragments['PrecursorMz'].tolist()[:1]
pro = fragments['ProductMz'].tolist()[:6]
peptide = q.scores.loc[q.scores.peptide_id == peptide_id, 'FullPeptideName'].iloc[0]
charge = q.scores.loc[q.scores.peptide_id == peptide_id, 'Charge'].iloc[0]
rank = q.scores.loc[q.scores.peptide_id == peptide_id, 'peak_group_rank'].iloc[0]
rt = q.scores.loc[q.scores.peptide_id == peptide_id, 'RT'].iloc[0]
print(f'ms2_name = \'{ms2_name}\'')
print(f'peptide, charge, rank, rt, pre = \'{peptide}\', {charge}, {rank}, {rt}, {pre[0]}')
print(f'pro = {pro}')
assert len(pre) > 0
assert len(pro) > 0
peak_group = requant.RequantPeakGroup(peptide, charge, rank, rt, pre[0], pro)
print(operator.run(ms2_name, [peak_group]))
Quantify & visualise the high scoring peptides¶
SGS data¶
[17]:
run_python_requant(
file=sgs_file,
quantifiers=sgs_quantifiers,
peptide_ids=sgs_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-008'
peptide, charge, rank, rt, pre = 'IVDVDLTSEGK(UniMod:259)', 2, 1, 2962.99, 592.318
pro = [642.355, 757.382, 856.45, 971.477]
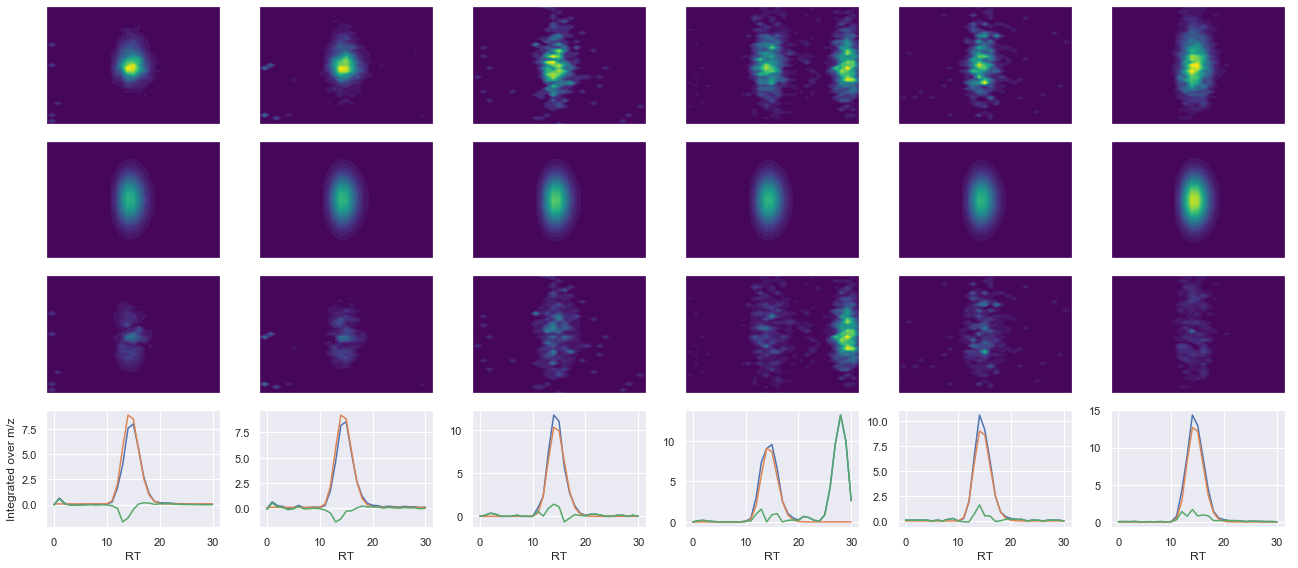
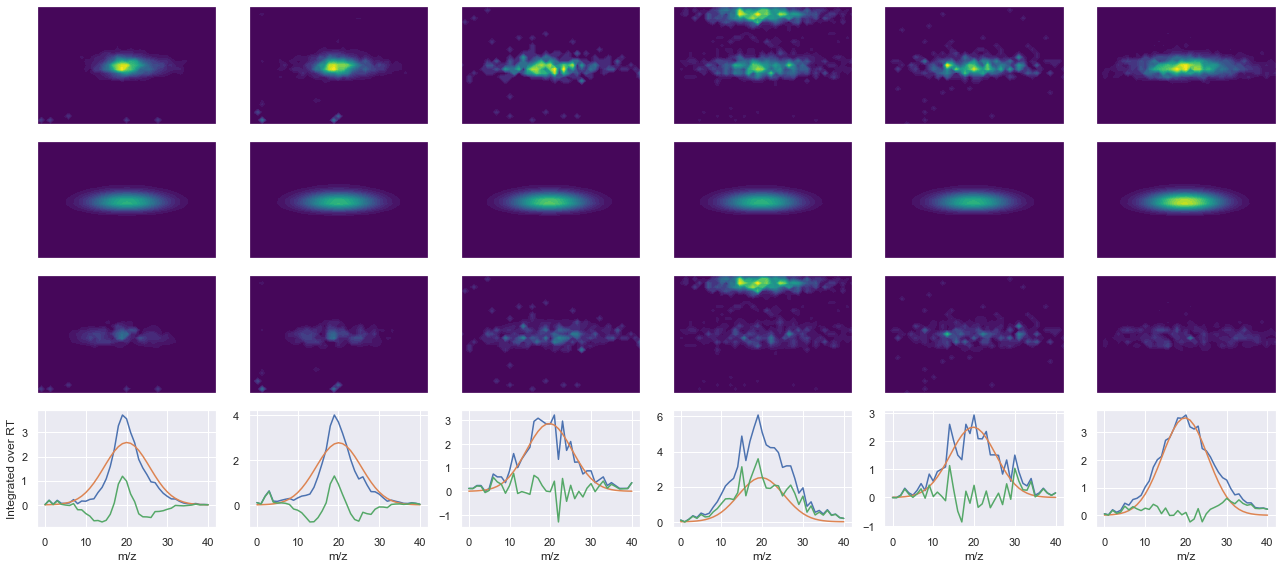
Raw MS1 & MS2: 1901.0151427865394 832.5617156912243
Fit MS1 & MS2: 1913.1799130745012 614.3915440003645
[('IVDVDLTSEGK(UniMod:259)', 2, 1913.1799130745012, 614.3915440003645)]
--------------------------------------------------
ms2_name = 'ms2-008'
peptide, charge, rank, rt, pre = 'NPDGTVTVISR(UniMod:267)', 2, 1, 2360.46, 584.813
pro = [484.312, 684.428, 842.497, 957.524]
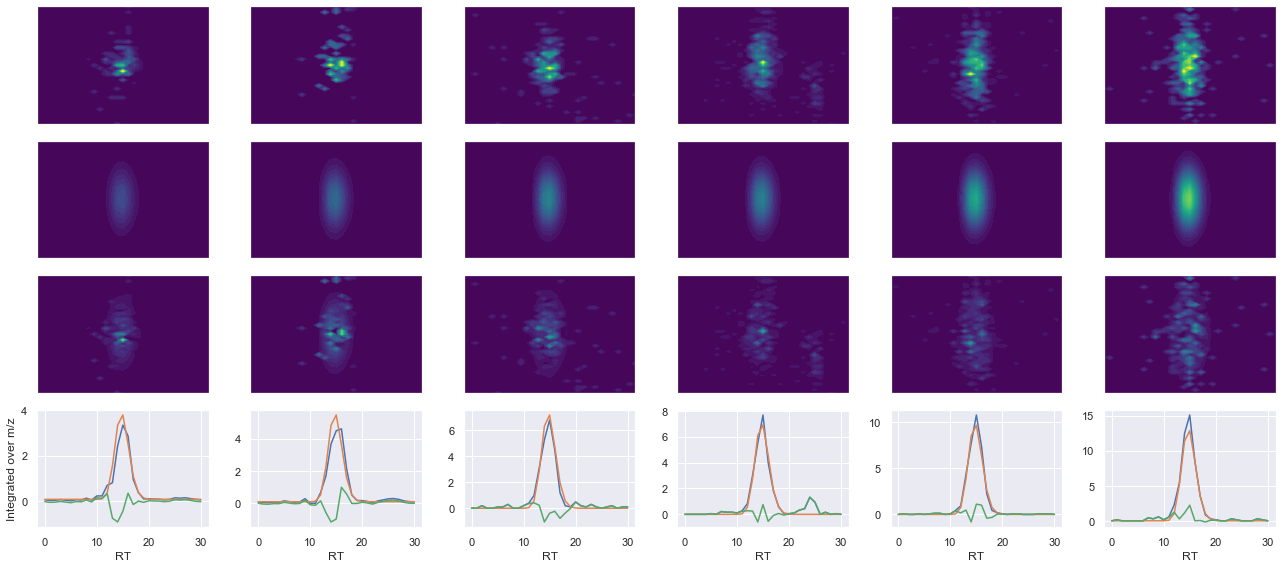
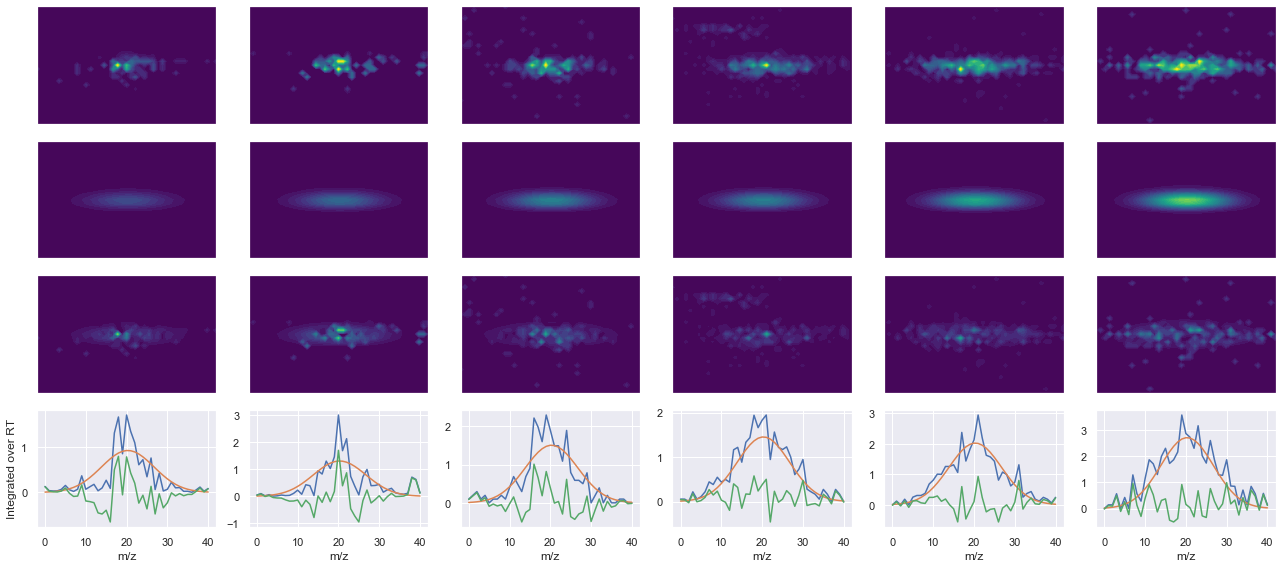
Raw MS1 & MS2: 1068.797569083983 226.6386387566327
Fit MS1 & MS2: 964.1610009504825 201.86531477130598
[('NPDGTVTVISR(UniMod:267)', 2, 964.1610009504825, 201.86531477130598)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-010'
peptide, charge, rank, rt, pre = 'IVEIYGPESSGK(UniMod:259)', 2, 1, 2900.32, 643.84
pro = [669.329, 832.393, 945.477, 1074.52]
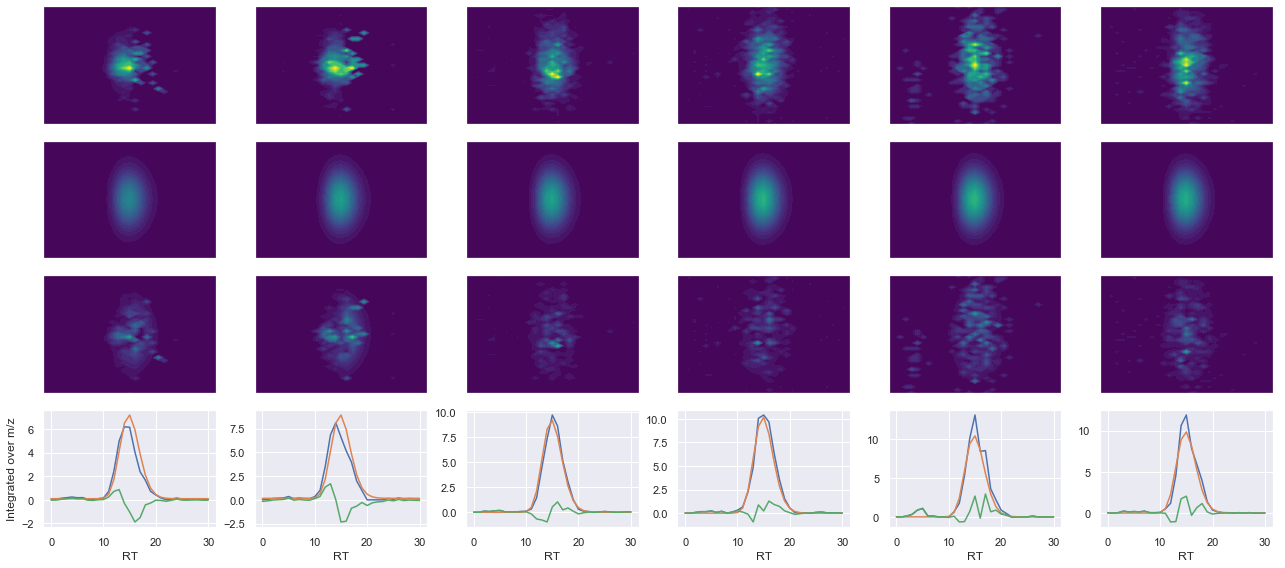
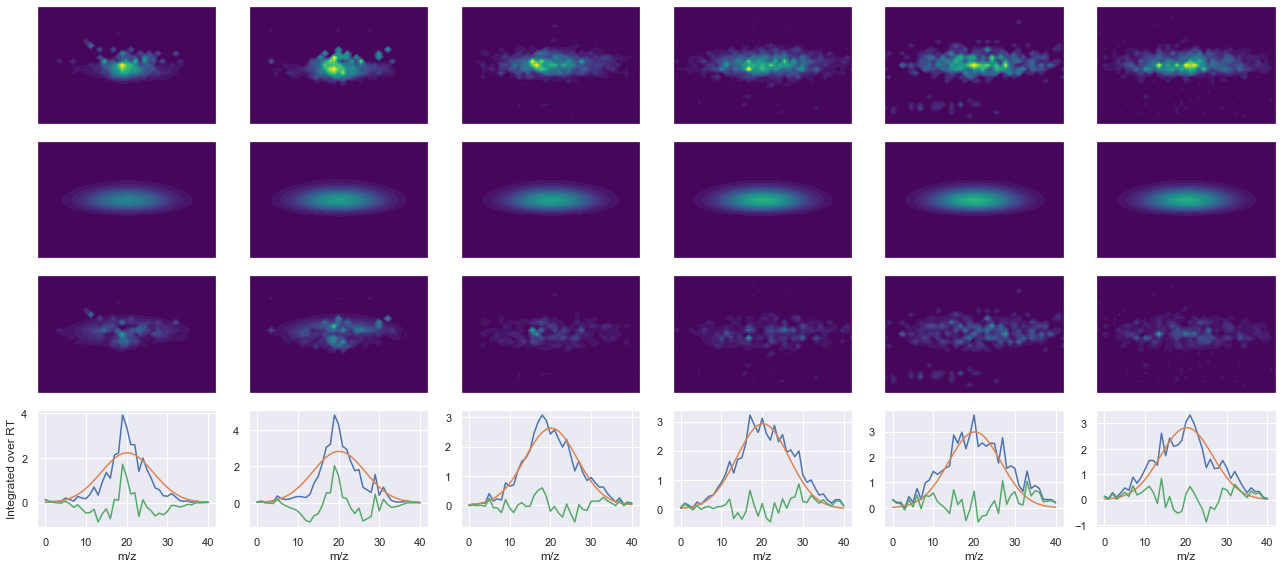
Raw MS1 & MS2: 1343.0514804124023 587.4011974819572
Fit MS1 & MS2: 1361.4908623989802 540.0065696055367
[('IVEIYGPESSGK(UniMod:259)', 2, 1361.4908623989802, 540.0065696055367)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-014'
peptide, charge, rank, rt, pre = 'SDSSDTPPLPSPPGK(UniMod:259)', 2, 1, 2804.66, 745.367
pro = [800.476, 897.528, 998.576, 1113.6]
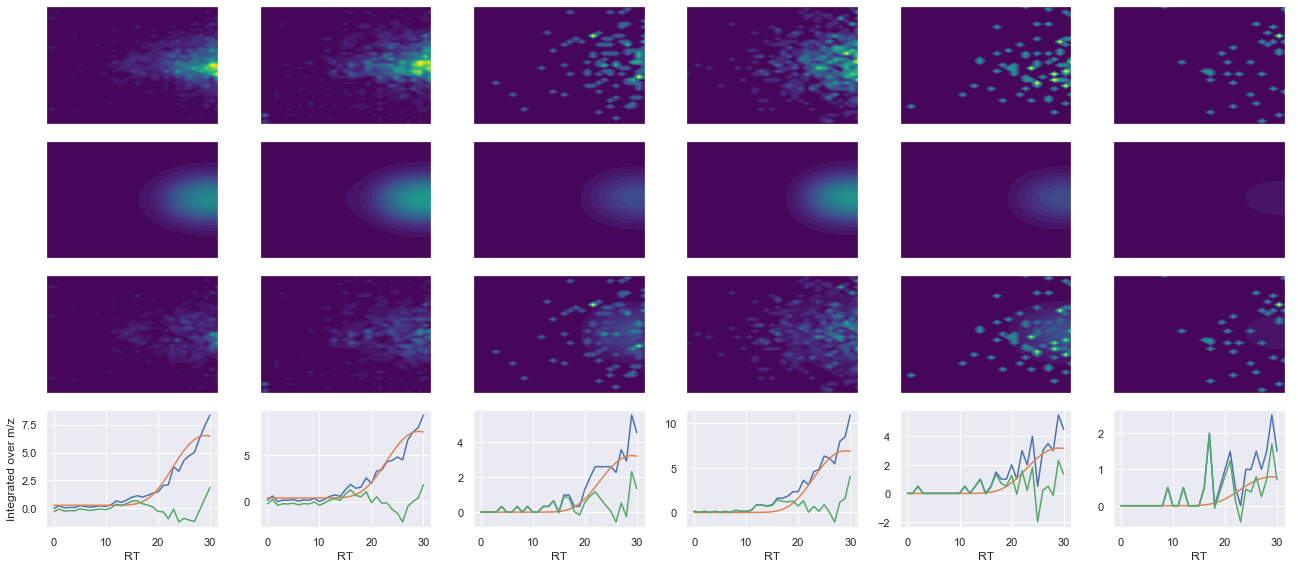
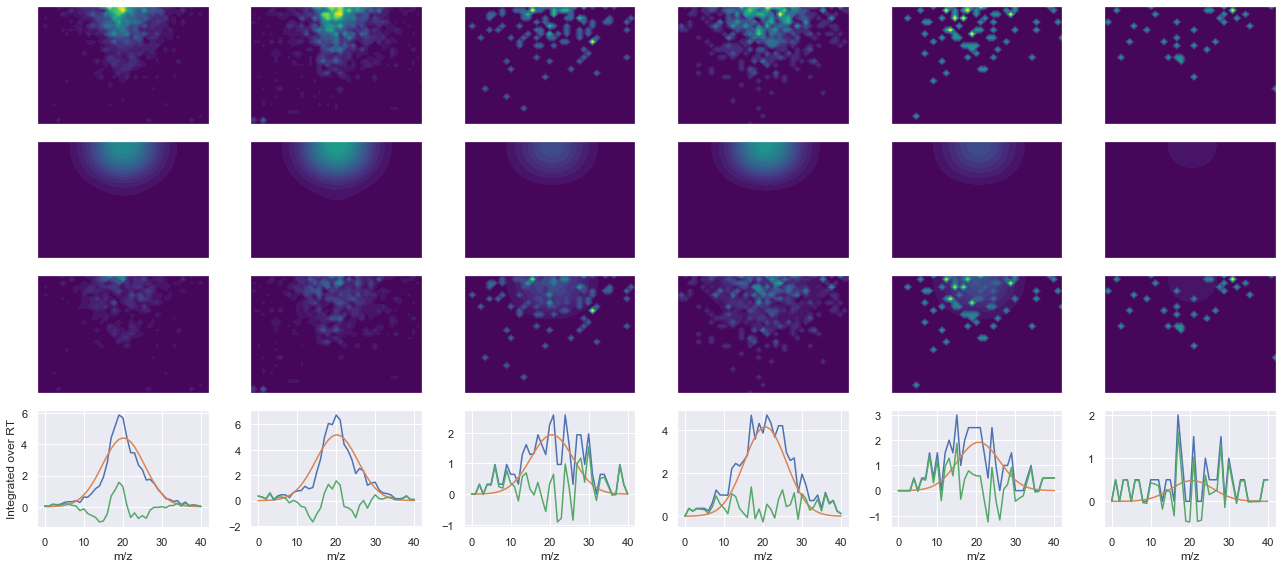
Raw MS1 & MS2: 592.4653454073219 122.54244489367036
Fit MS1 & MS2: 505.67140106941656 88.20516067864703
[('SDSSDTPPLPSPPGK(UniMod:259)', 2, 505.67140106941656, 88.20516067864703)]
ProCan90 data¶
[18]:
run_python_requant(
file=procan90_file,
quantifiers=procan90_quantifiers,
peptide_ids=procan90_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-024'
peptide, charge, rank, rt, pre = 'VSFELFADK', 2, 1, 3025.44, 528.27404785
pro = [435.22381592, 480.24526978, 593.3293457, 722.37194824, 869.44036865, 956.47235107]
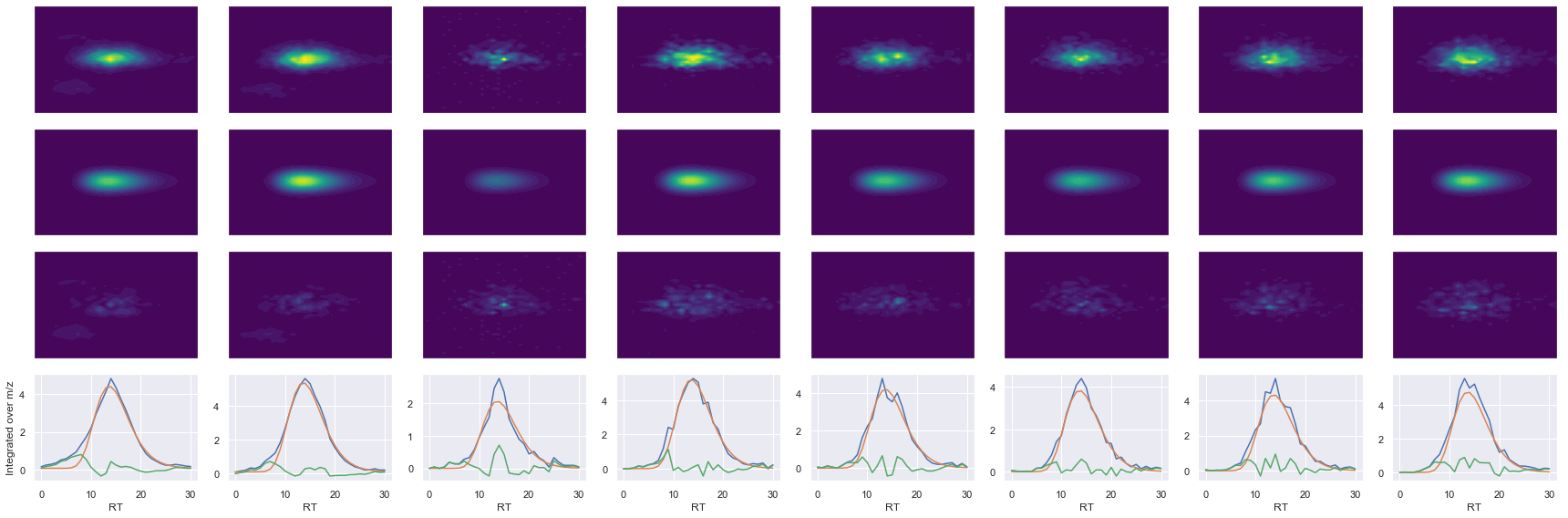
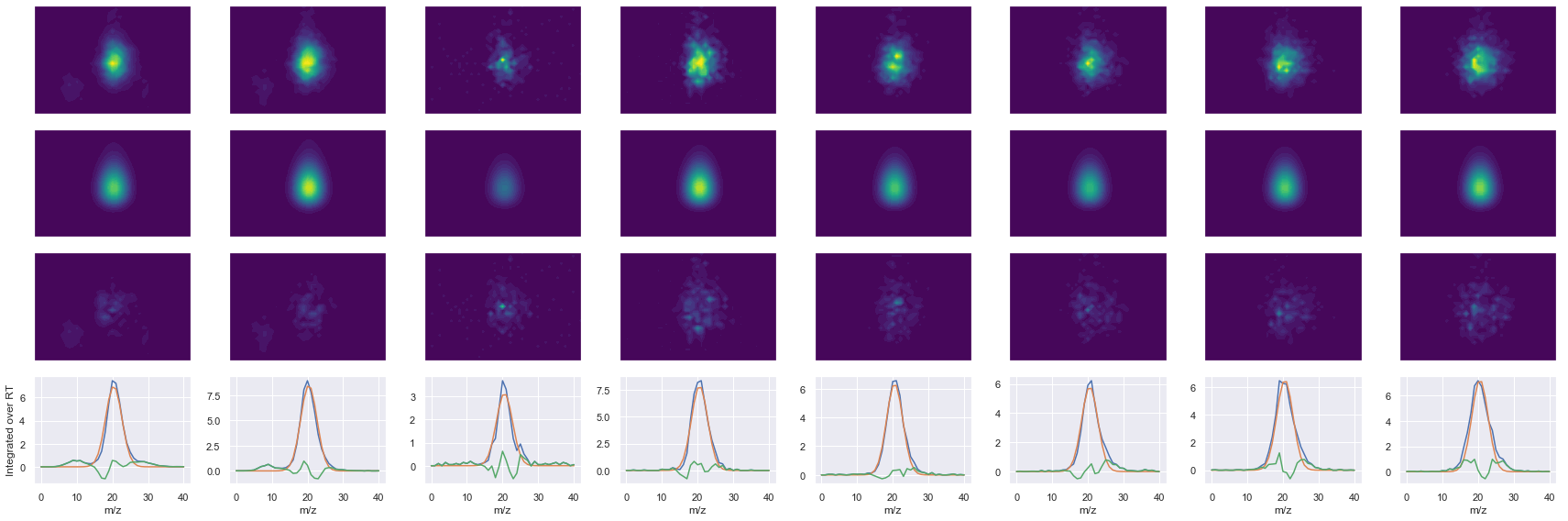
Raw MS1 & MS2: 28778.380265487547 6040.770575850728
Fit MS1 & MS2: 24396.972393554213 5284.702891876719
[('VSFELFADK', 2, 24396.972393554213, 5284.702891876719)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-035'
peptide, charge, rank, rt, pre = 'AIPQLQGYLR', 2, 1, 2607.96, 579.83514404
pro = [487.77453613, 508.28781128, 636.34637451, 749.43041992, 877.48901367, 974.54180908]
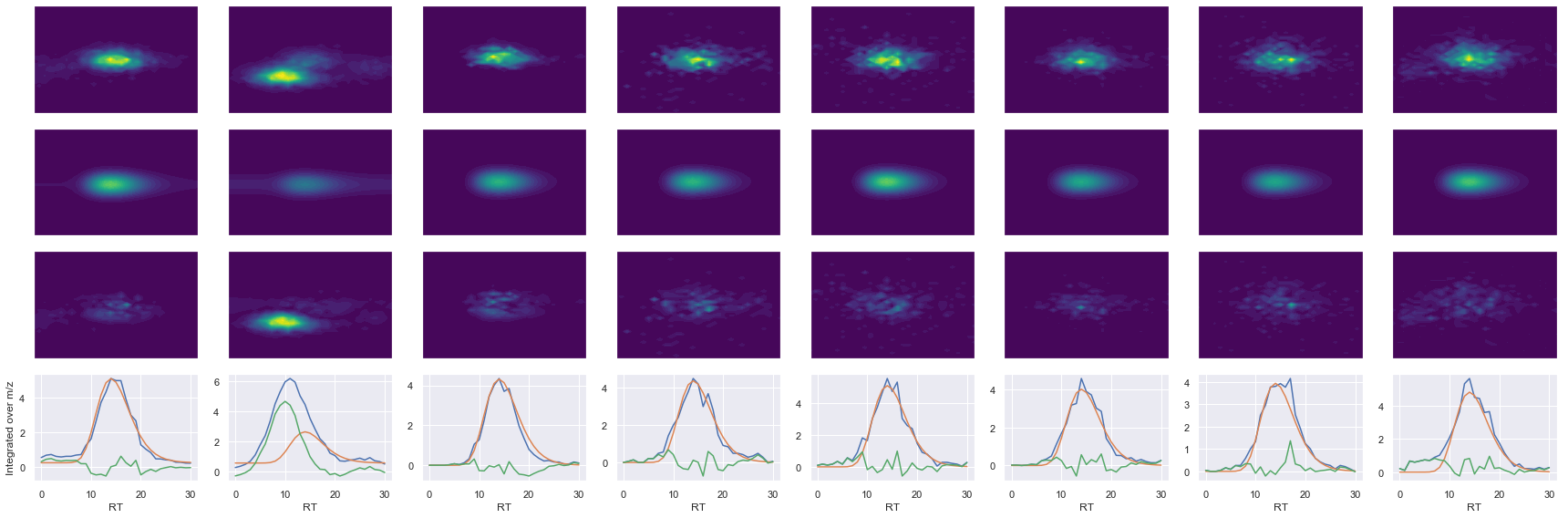
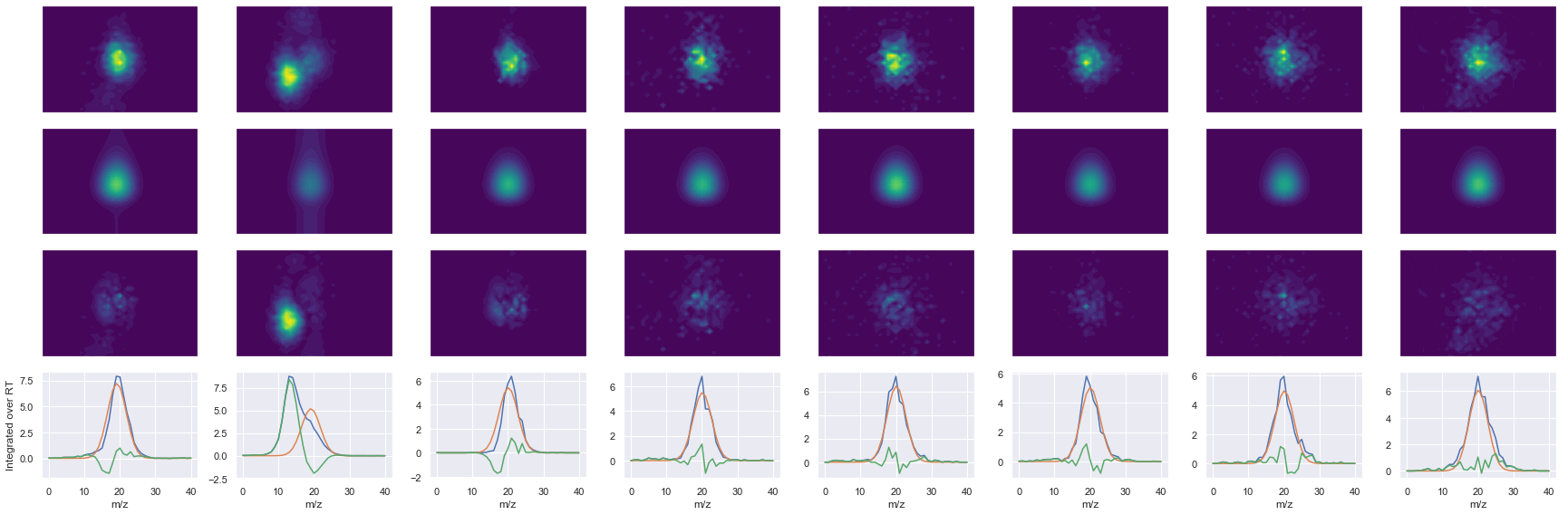
Raw MS1 & MS2: 30576.567646871867 4026.2126532143407
Fit MS1 & MS2: 12613.59797450274 3776.8008016967888
[('AIPQLQGYLR', 2, 12613.59797450274, 3776.8008016967888)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-048'
peptide, charge, rank, rt, pre = 'NALESYAFNMK', 2, 1, 2491.33, 644.30554199
pro = [539.26464844, 610.30175781, 773.36505127, 860.39709473, 989.43969727, 1102.5238037]
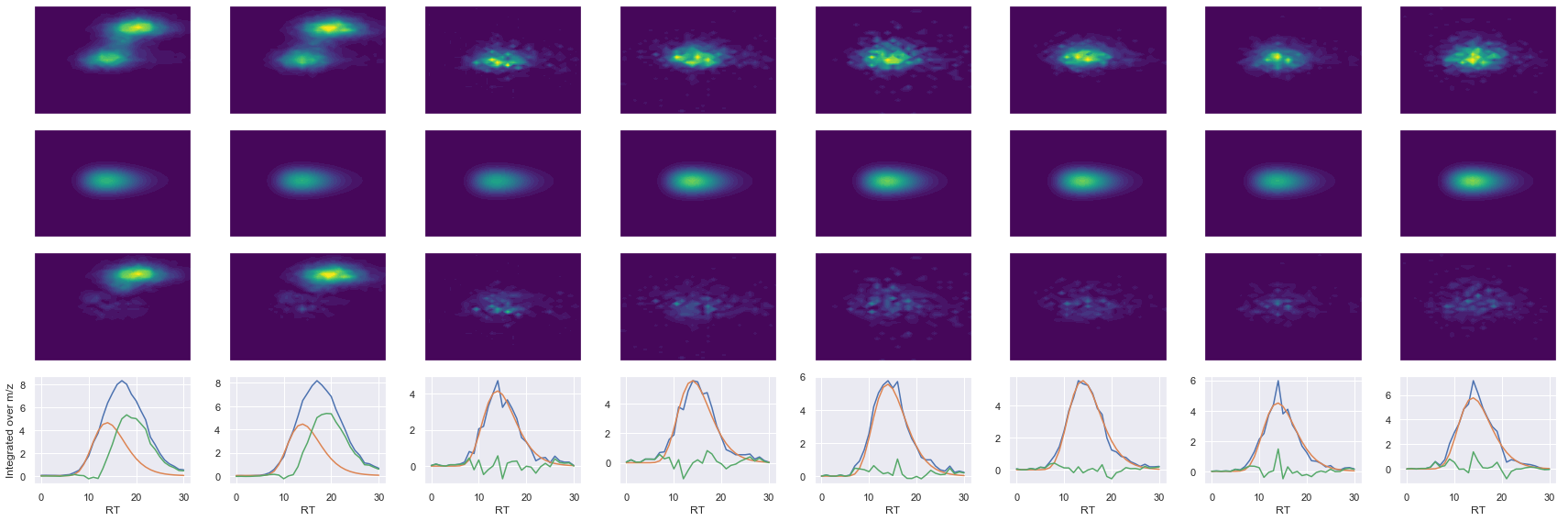
Raw MS1 & MS2: 69309.96573677666 4298.2830446508515
Fit MS1 & MS2: 29909.713574711433 4079.6536675590323
[('NALESYAFNMK', 2, 29909.713574711433, 4079.6536675590323)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-049'
peptide, charge, rank, rt, pre = 'VIHDNFGIVEGLMTTVHAITATQK', 4, 1, 4158.86, 649.59545898
pro = [548.30383301, 579.28851318, 732.42504883, 783.37841797, 869.48394775, 896.46246338]
Raw MS1 & MS2: 36075.46972826726 4581.8361977096065
Fit MS1 & MS2: 31349.392247620683 3715.5025255865135
[('VIHDNFGIVEGLMTTVHAITATQK', 4, 31349.392247620683, 3715.5025255865135)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-055'
peptide, charge, rank, rt, pre = 'GGGGNFGPGPGSNFR', 2, 1, 1180.7, 689.31835938
pro = [580.28381348, 677.33654785, 734.35803223, 831.4107666, 888.43225098, 1035.5006104]
Raw MS1 & MS2: 18984.09727504625 3965.848553410137
Fit MS1 & MS2: 15462.107708268175 3531.5971517279813
[('GGGGNFGPGPGSNFR', 2, 15462.107708268175, 3531.5971517279813)]
Quantify & visualise the outlier peptides¶
SGS data¶
[19]:
run_python_requant(
file=sgs_file,
quantifiers=sgs_quantifiers,
peptide_ids=sgs_outlier_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-011'
peptide, charge, rank, rt, pre = 'YYDYTLSINGK(UniMod:259)', 2, 1, 3657.93, 672.832
pro = [526.307, 639.392, 740.439, 1018.53]
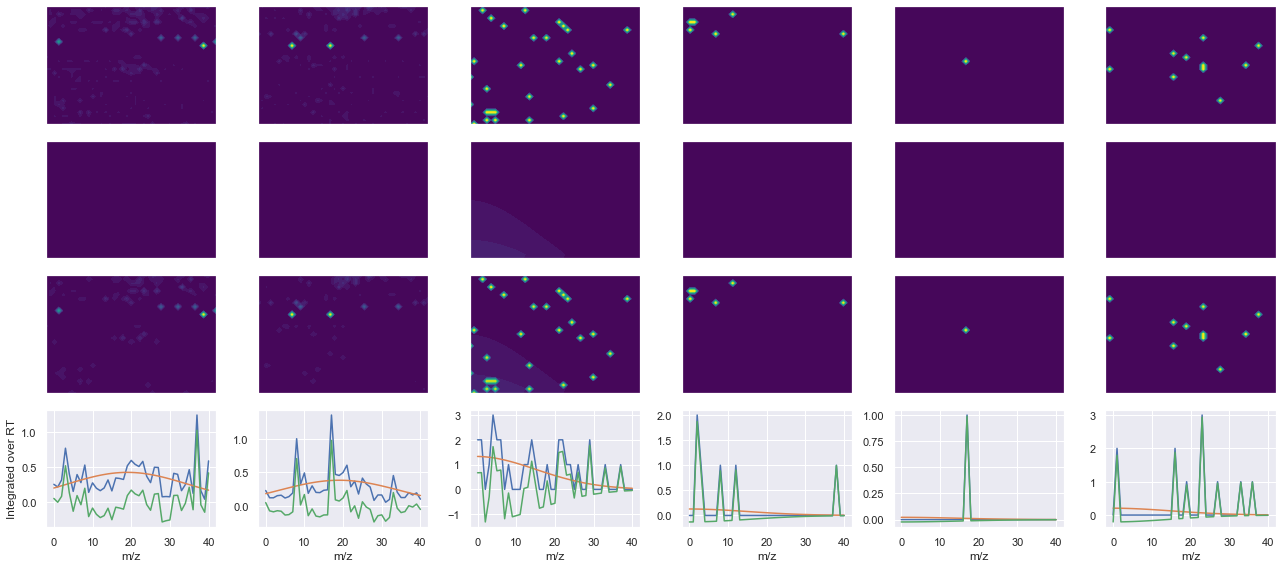
Raw MS1 & MS2: 93.2215802795607 5.8908822504070155
Fit MS1 & MS2: 6.277257202415132e-06 3.8327705199061106
[('YYDYTLSINGK(UniMod:259)', 2, 6.277257202415132e-06, 3.8327705199061106)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-012'
peptide, charge, rank, rt, pre = 'TIVGVSEDFAALR(UniMod:267)', 2, 1, 4574.05, 694.376
pro = [587.354, 918.455, 1074.55, 1173.61]
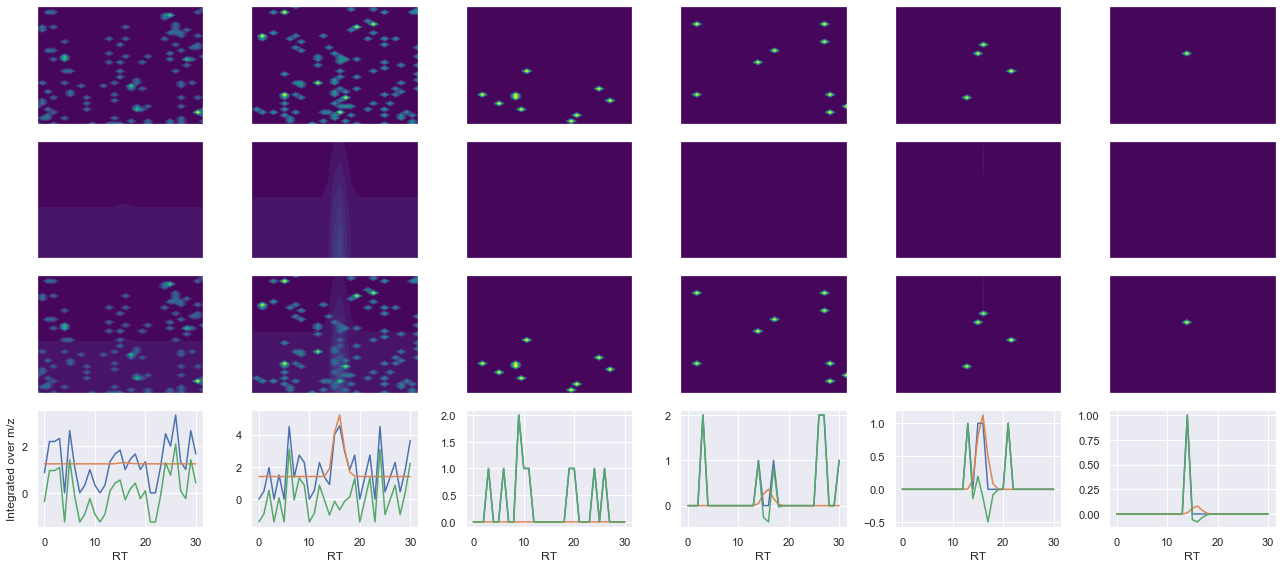
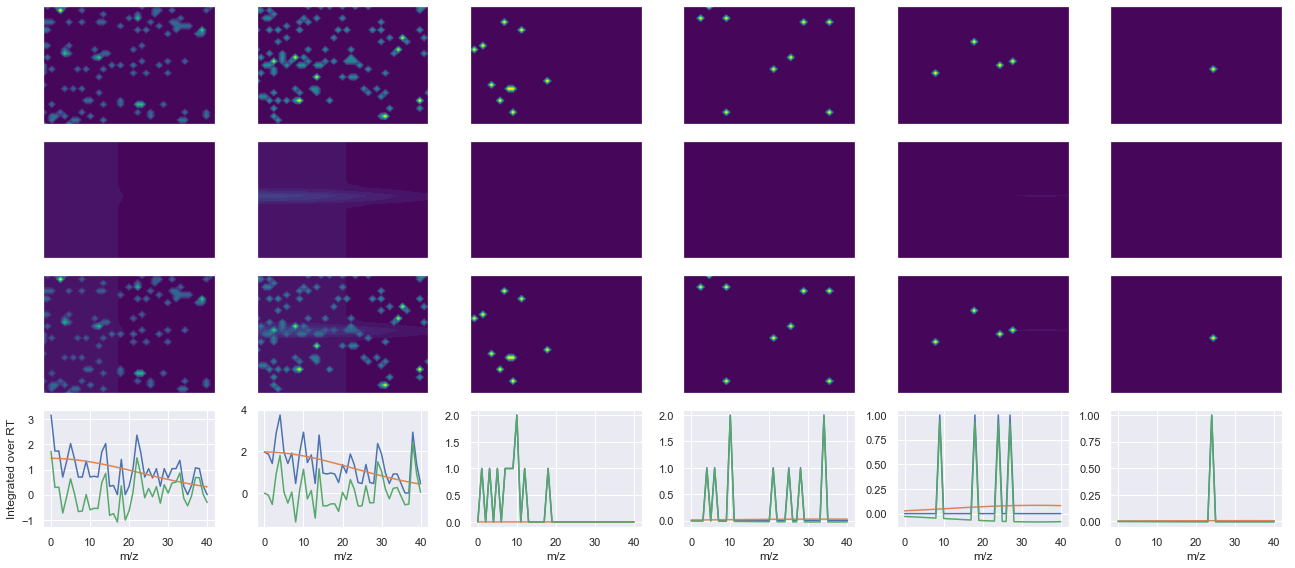
Raw MS1 & MS2: 30.68848719714169 3.290575667970753
Fit MS1 & MS2: 2.5708773559614384 0.5779817408456864
[('TIVGVSEDFAALR(UniMod:267)', 2, 2.5708773559614384, 0.5779817408456864)]
--------------------------------------------------
ms2_name = 'ms2-012'
peptide, charge, rank, rt, pre = 'VESPVLPPVLVPK(UniMod:259)', 2, 1, 4738.54, 691.431
pro = [757.506, 870.59, 1066.71, 1153.74]
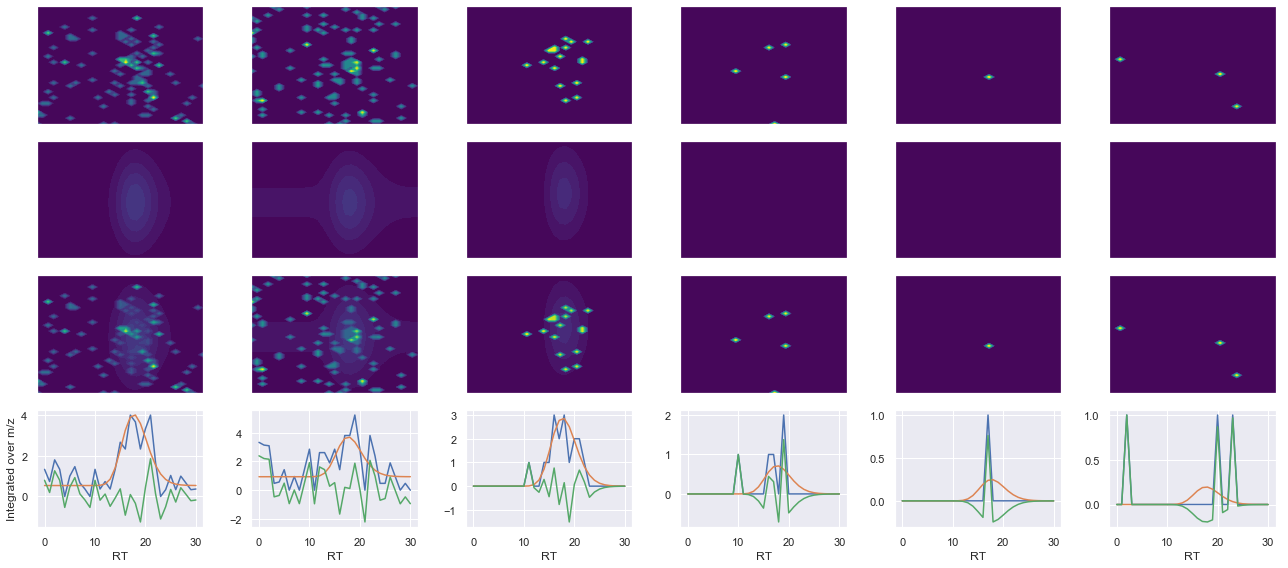
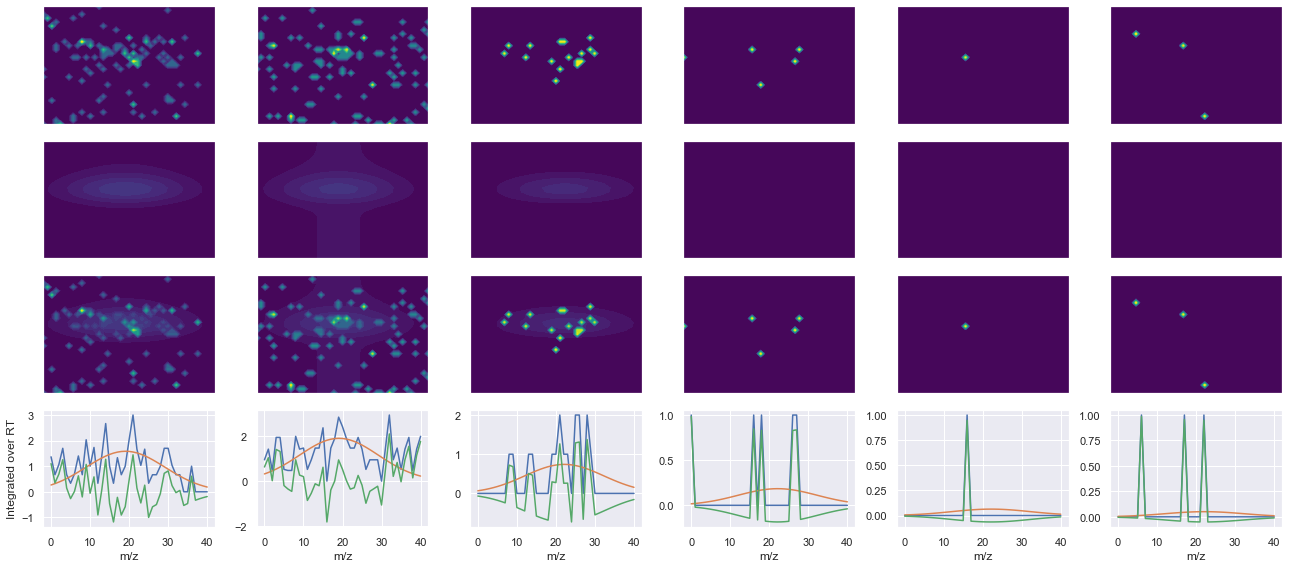
Raw MS1 & MS2: 30.267127515105777 3.626600767829161
Fit MS1 & MS2: 12.886623304127127 3.457505682641535
[('VESPVLPPVLVPK(UniMod:259)', 2, 12.886623304127127, 3.457505682641535)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-014'
peptide, charge, rank, rt, pre = 'EYTEGVNGQPSIR(UniMod:267)', 2, 1, 2438.35, 730.356
pro = [667.376, 781.419, 937.509, 1066.55]
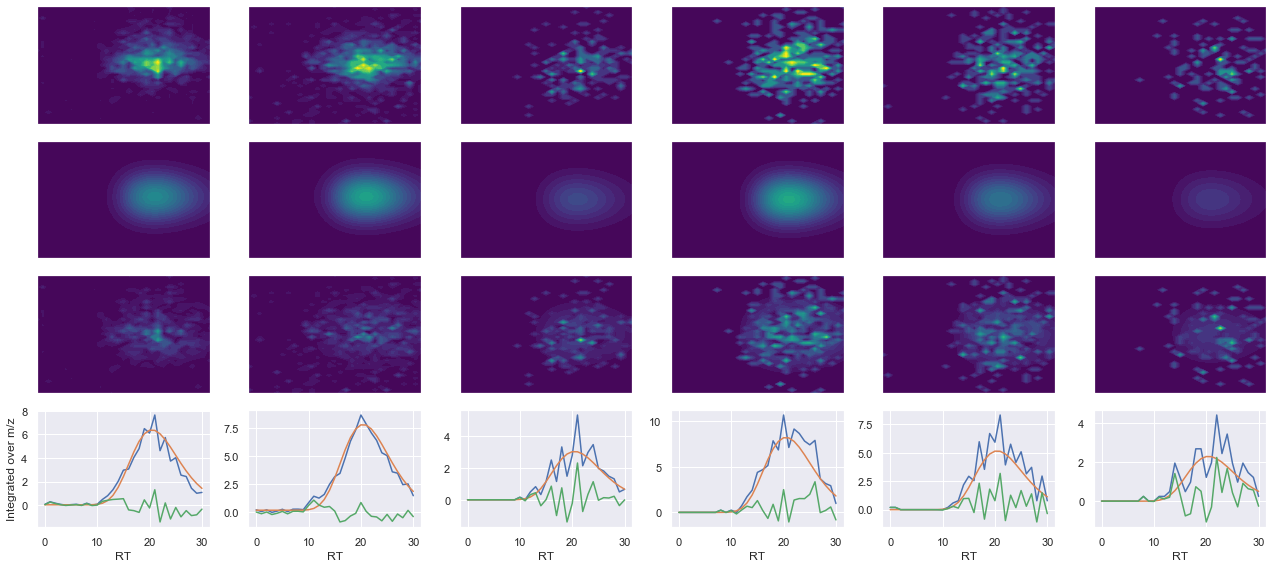
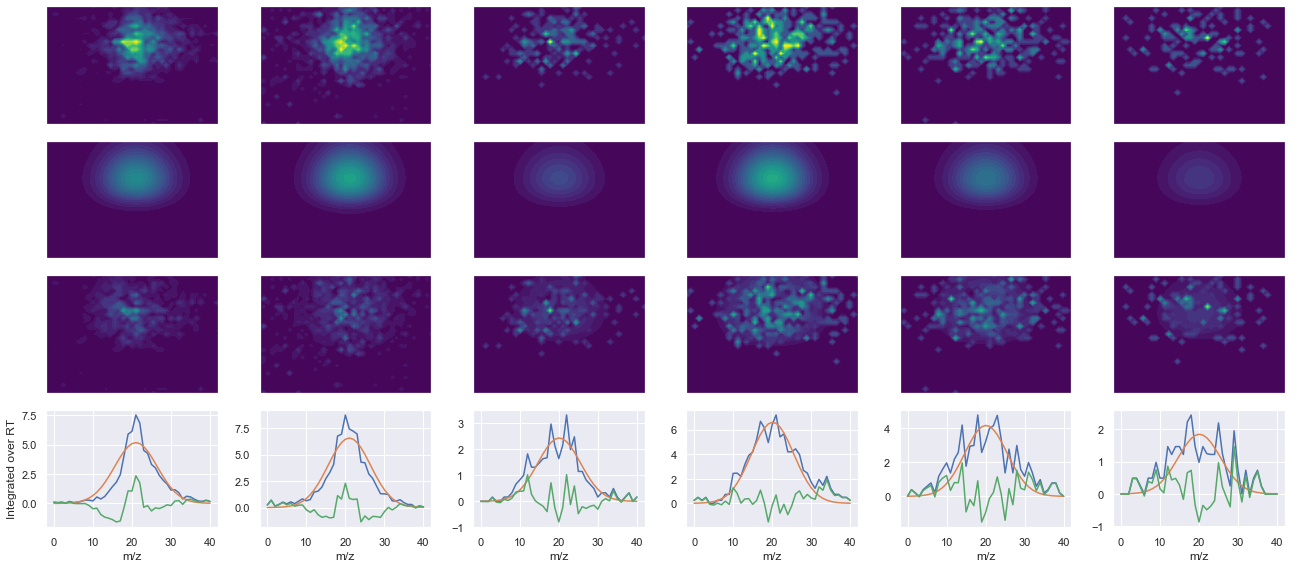
Raw MS1 & MS2: 571.6679698524539 159.05646570609215
Fit MS1 & MS2: 569.5681733014899 134.3360836457496
[('EYTEGVNGQPSIR(UniMod:267)', 2, 569.5681733014899, 134.3360836457496)]
--------------------------------------------------
ms2_name = 'ms2-014'
peptide, charge, rank, rt, pre = 'MIVDPVEPHGEMK(UniMod:259)', 2, 1, 2851.66, 745.367
pro = [706.343, 835.386, 1031.51, 1245.6]
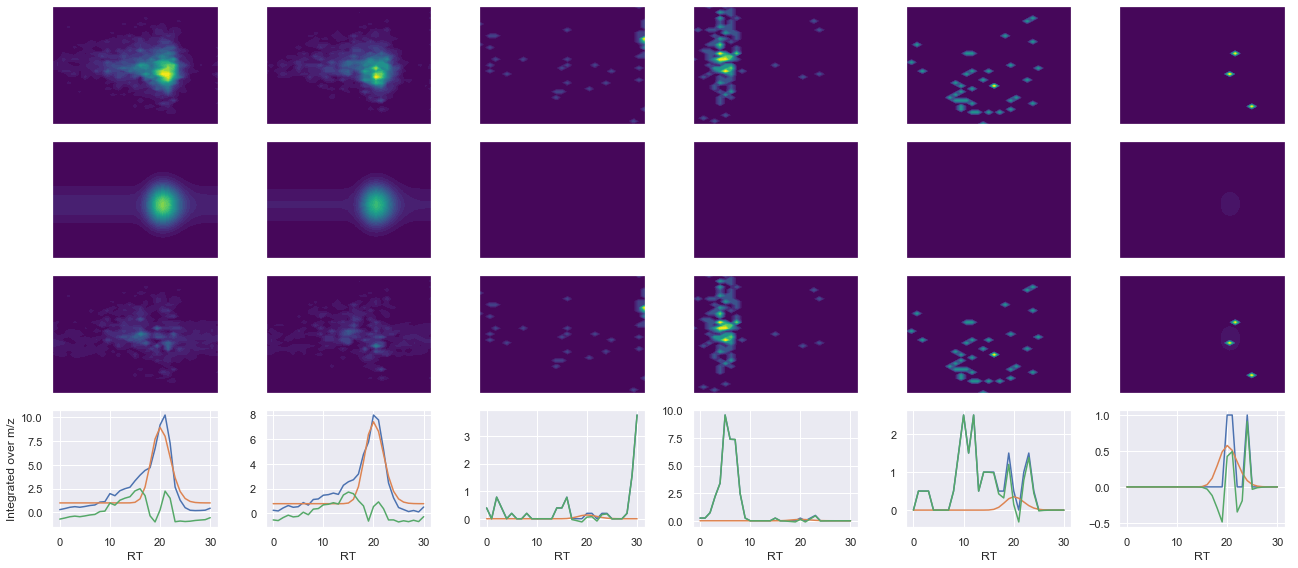
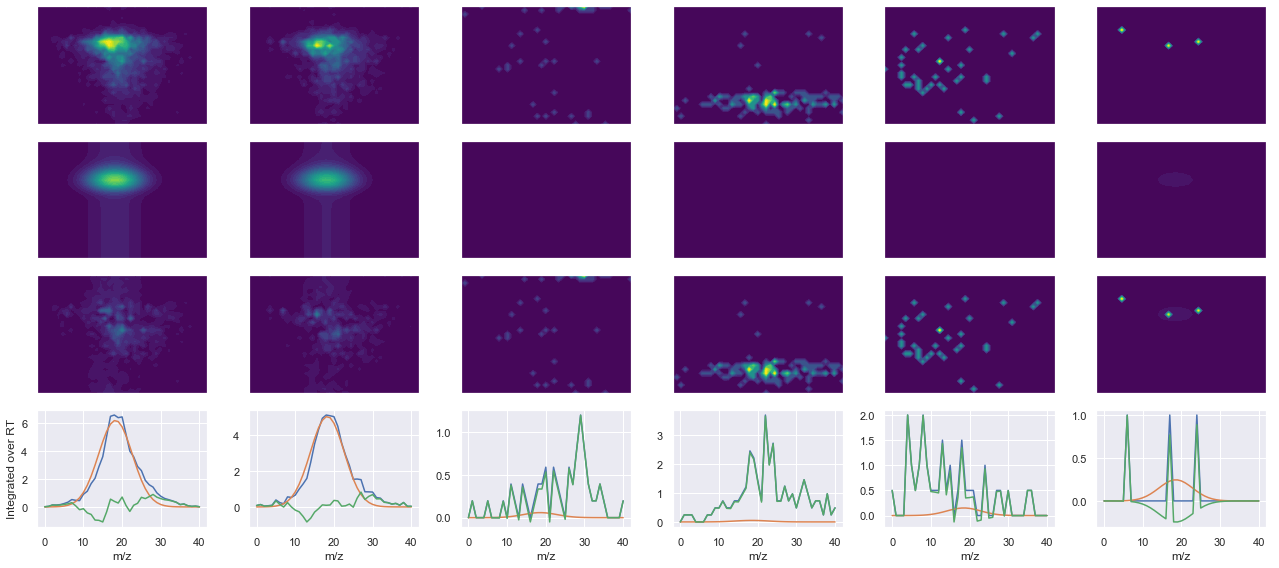
Raw MS1 & MS2: 1431.3119159291473 33.13252129485163
Fit MS1 & MS2: 732.8624744794847 1.724481319894012
[('MIVDPVEPHGEMK(UniMod:259)', 2, 732.8624744794847, 1.724481319894012)]
--------------------------------------------------
ms2_name = 'ms2-014'
peptide, charge, rank, rt, pre = 'SDSSDTPPLPSPPGK(UniMod:259)', 2, 1, 2804.66, 745.367
pro = [800.476, 897.528, 998.576, 1113.6]
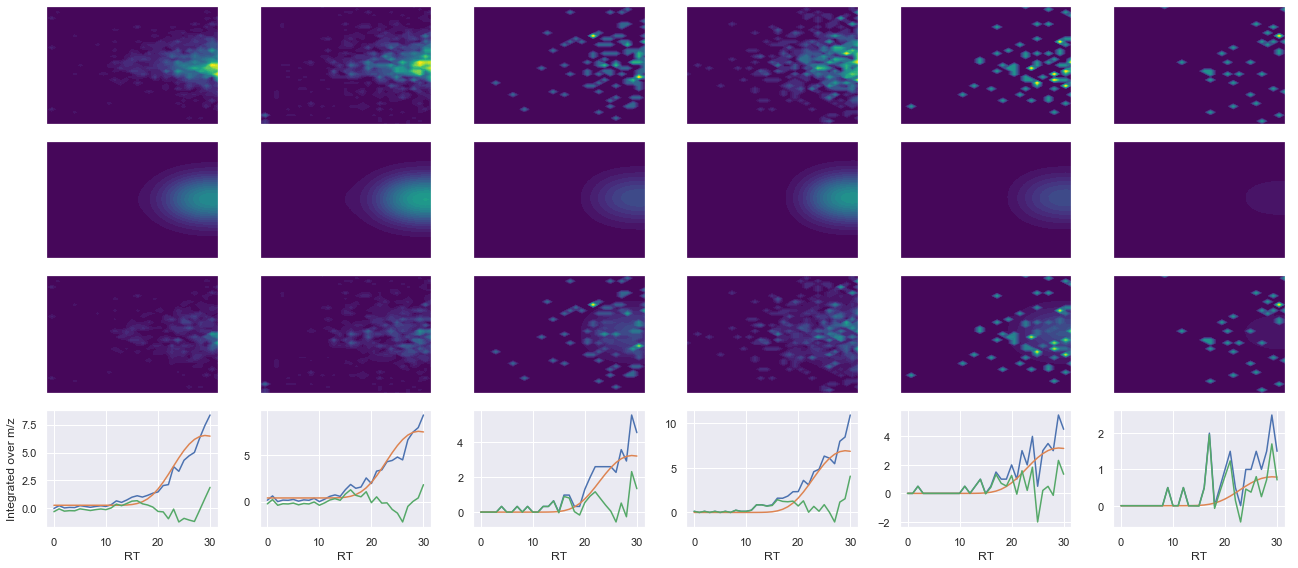
Raw MS1 & MS2: 592.4653454073219 122.54244489367036
Fit MS1 & MS2: 505.67140106941656 88.20516067864703
[('SDSSDTPPLPSPPGK(UniMod:259)', 2, 505.67140106941656, 88.20516067864703)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-015'
peptide, charge, rank, rt, pre = 'FPSPVSHADDLYGK(UniMod:259)', 2, 1, 3293.18, 770.88
pro = [789.387, 1013.48, 1209.6, 1296.63]
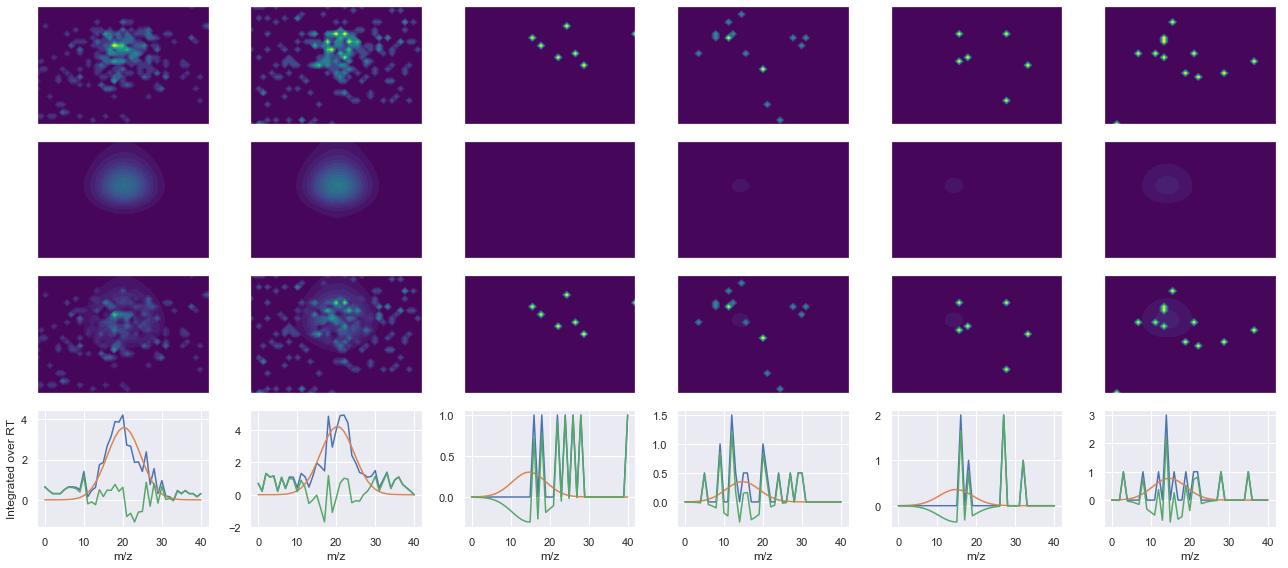
Raw MS1 & MS2: 87.85798084824421 6.512684995738631
Fit MS1 & MS2: 56.37263352773063 3.721348294688908
[('FPSPVSHADDLYGK(UniMod:259)', 2, 56.37263352773063, 3.721348294688908)]
--------------------------------------------------
ms2_name = 'ms2-015'
peptide, charge, rank, rt, pre = 'NTDFNGVNNIHQK(UniMod:259)', 2, 1, 2128.47, 754.87
pro = [647.371, 917.504, 1031.55, 1178.62]
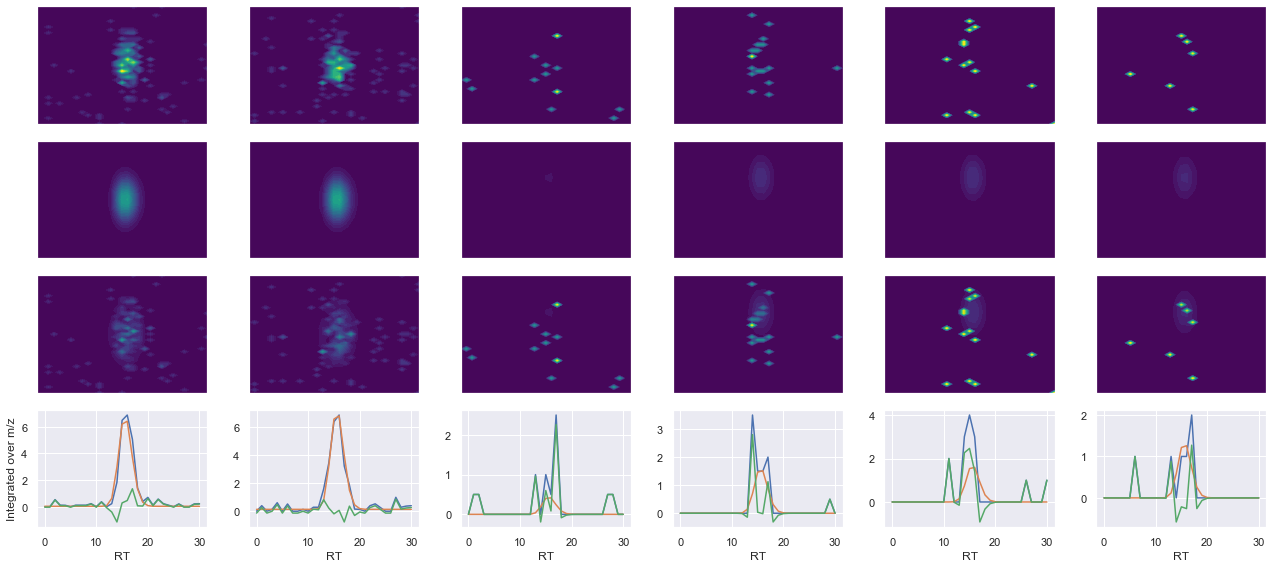
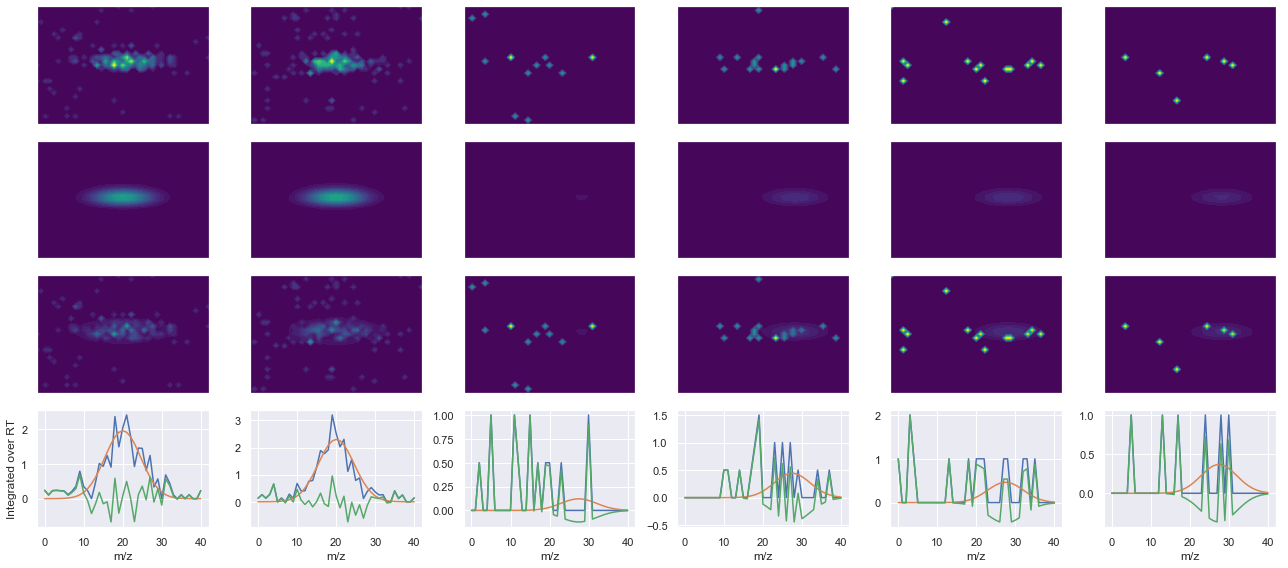
Raw MS1 & MS2: 77.90595348022188 7.52938575325957
Fit MS1 & MS2: 61.73288675398399 3.375192854570683
[('NTDFNGVNNIHQK(UniMod:259)', 2, 61.73288675398399, 3.375192854570683)]
ProCan90 data¶
[20]:
run_python_requant(
file=procan90_file,
quantifiers=procan90_quantifiers,
peptide_ids=procan90_outlier_peptide_ids,
)
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-004'
peptide, charge, rank, rt, pre = 'QNVPIIR', 2, 1, 1062.29, 420.2585144
pro = [401.28707886, 498.33984375, 597.40826416, 711.45117188]
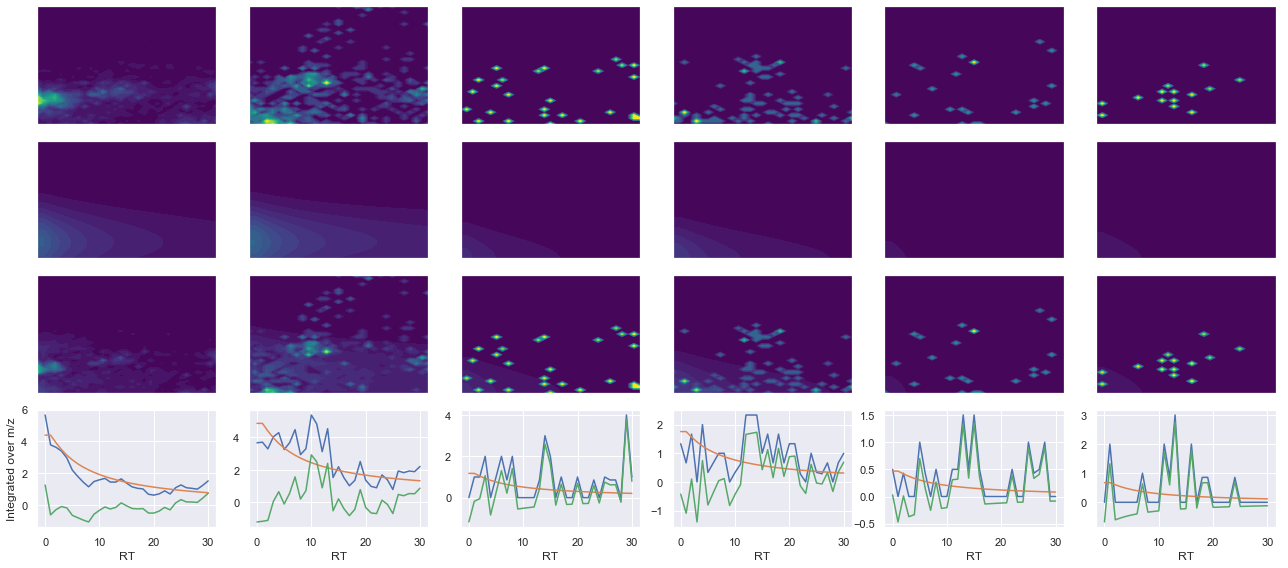
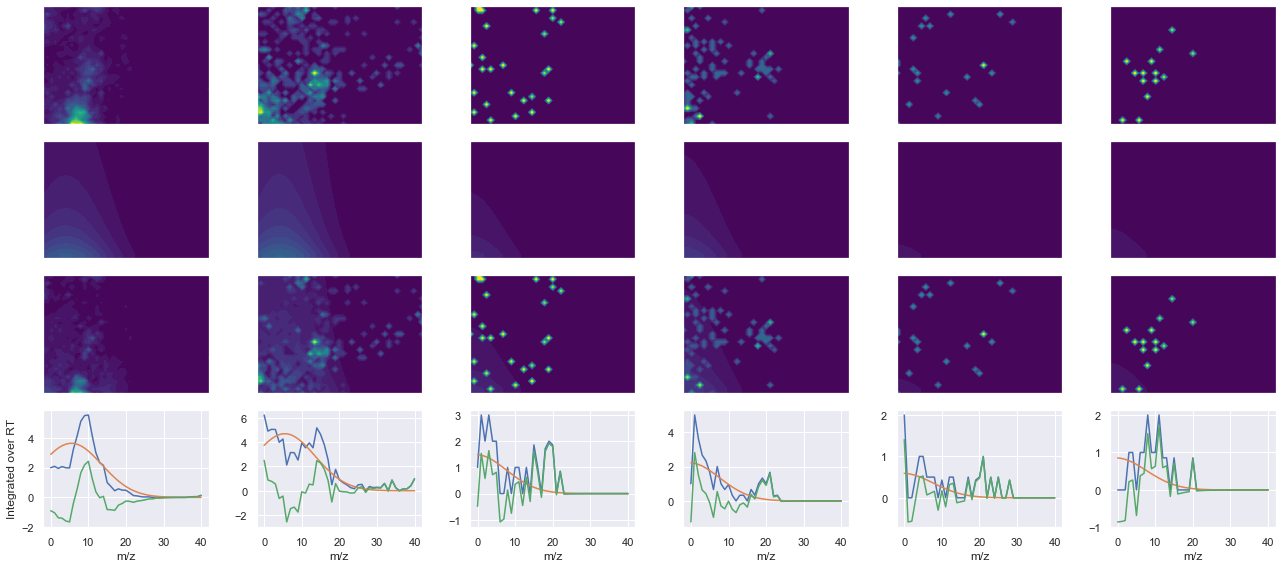
Raw MS1 & MS2: 2748.484368356817 66.35065408147446
Fit MS1 & MS2: 2799.4163691023036 45.93175784643054
[('QNVPIIR', 2, 2799.4163691023036, 45.93175784643054)]
--------------------------------------------------
ms2_name = 'ms2-004'
peptide, charge, rank, rt, pre = 'REDFLR', 2, 1, 1997.44, 418.22467041
pro = [435.27142334, 550.29840088, 661.3303833]
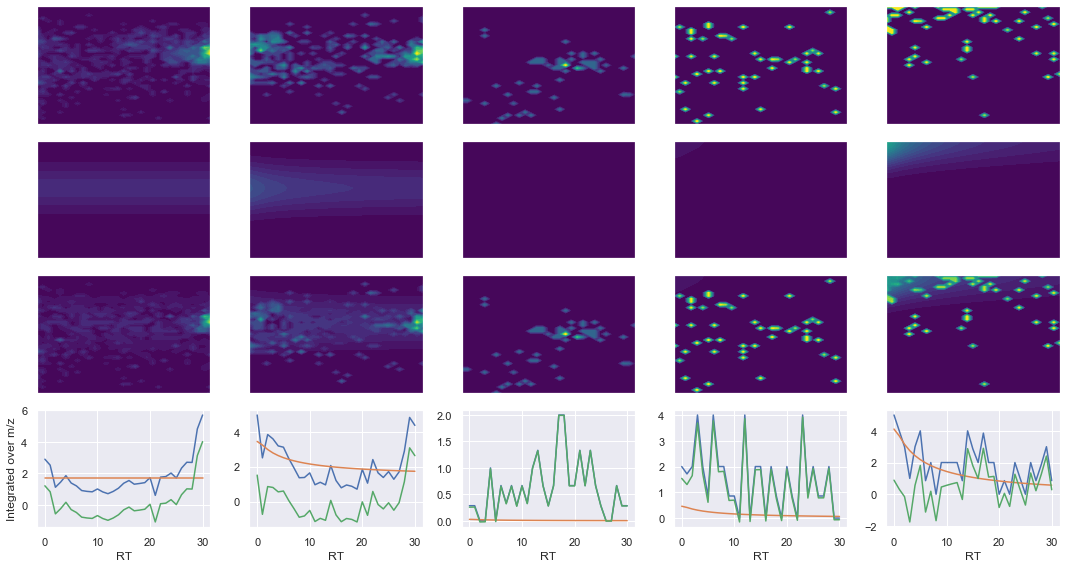
Raw MS1 & MS2: 1371.4563234616016 75.66820364750609
Fit MS1 & MS2: 148.42967820914942 24.819024099327837
[('REDFLR', 2, 148.42967820914942, 24.819024099327837)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-049'
peptide, charge, rank, rt, pre = 'HGEPEEDIVGLQAFQER', 3, 1, 2662.94, 651.98156738
pro = [579.28851318, 778.38421631, 794.29516602, 907.37921143, 948.48974609, 1063.4691162]
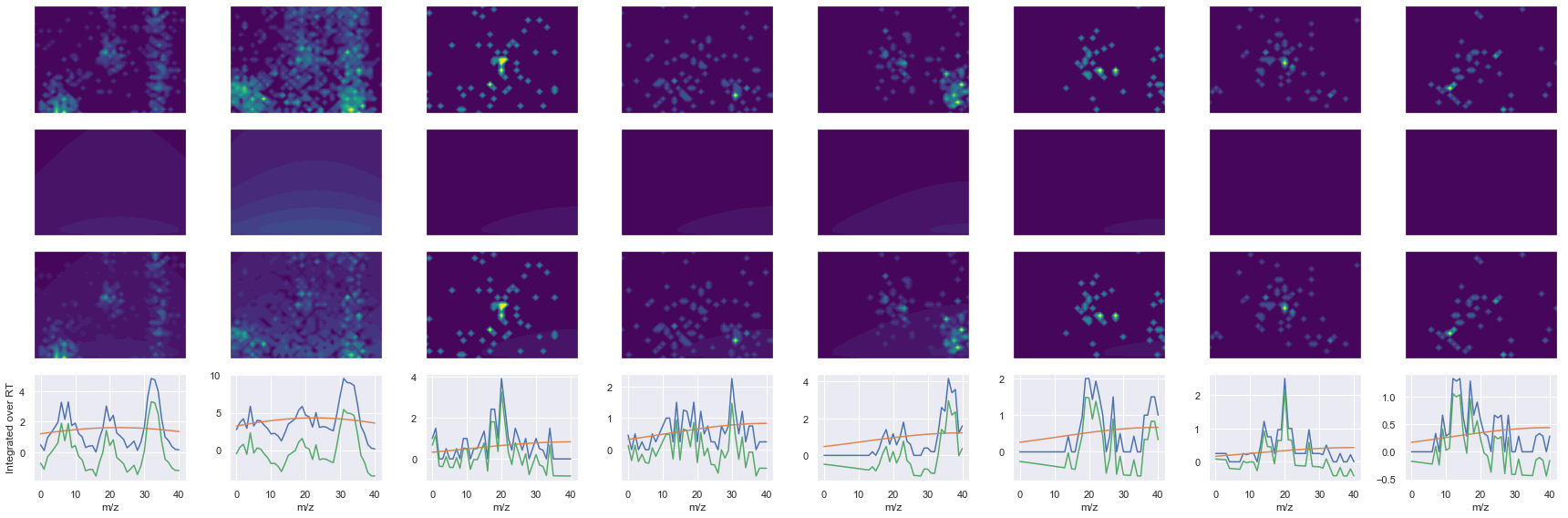
Raw MS1 & MS2: 2602.4031337189504 258.6497463384277
Fit MS1 & MS2: 1916.315200109147 267.09342144412926
[('HGEPEEDIVGLQAFQER', 3, 1916.315200109147, 267.09342144412926)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-050'
peptide, charge, rank, rt, pre = 'NADMQEELIASLEEQLK', 3, 1, 4136.85, 654.3225708
pro = [517.29803467, 759.42468262, 818.29852295, 846.45672607, 917.49383545, 931.38256836]
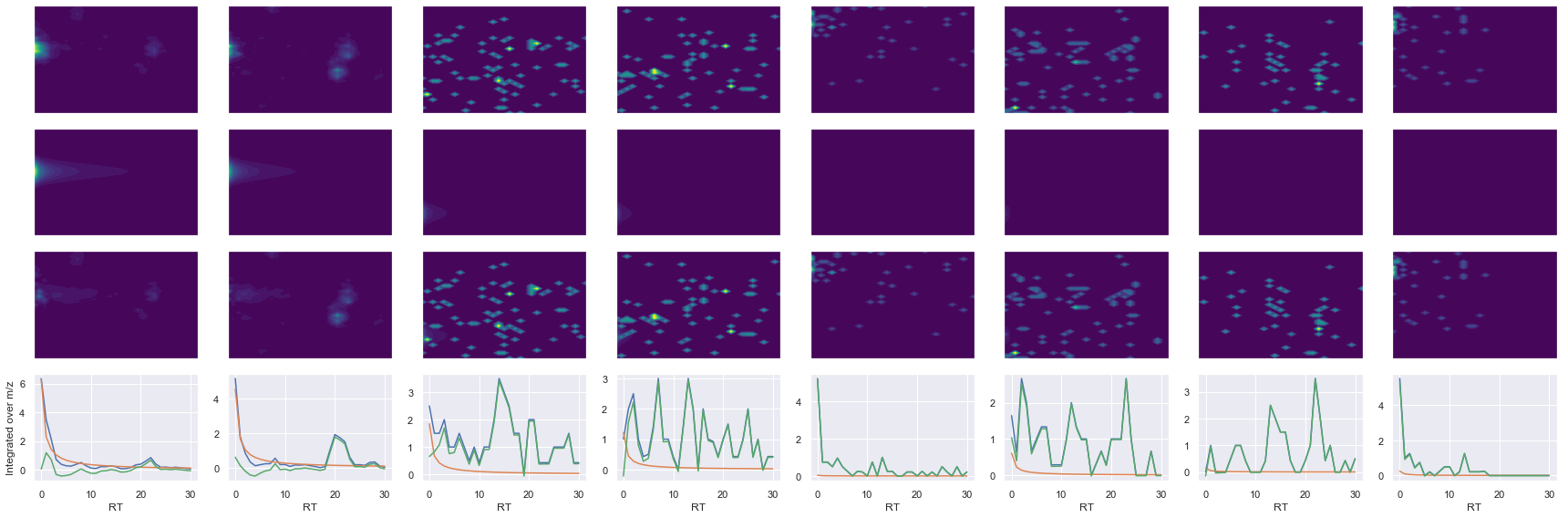
Raw MS1 & MS2: 11887.751376622578 206.13587926308224
Fit MS1 & MS2: 10432.259282198298 14.468248025629837
[('NADMQEELIASLEEQLK', 3, 10432.259282198298, 14.468248025629837)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-057'
peptide, charge, rank, rt, pre = 'VNITPAEVGVLVGK', 2, 1, 3101.9, 698.41394043
pro = [416.28674316, 525.30310059, 572.37664795, 871.52471924, 968.57751465, 1069.6252441]
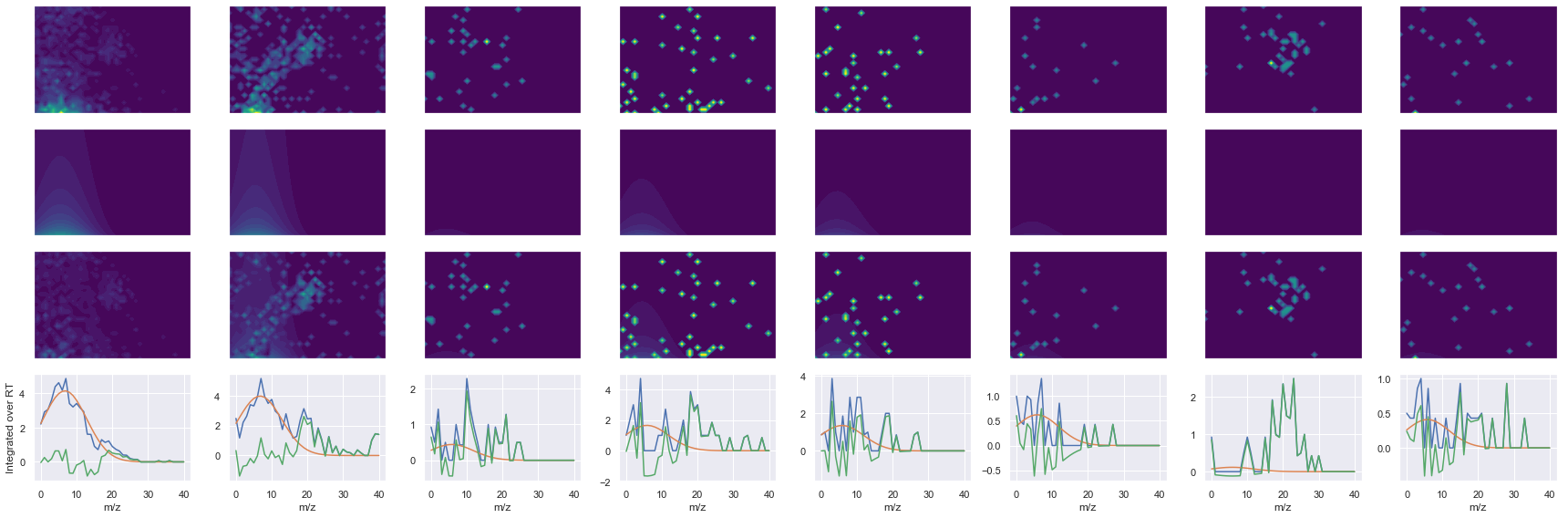
Raw MS1 & MS2: 1822.2111671039456 88.9707300880708
Fit MS1 & MS2: 1422.579423536166 40.31177107899468
[('VNITPAEVGVLVGK', 2, 1422.579423536166, 40.31177107899468)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-064'
peptide, charge, rank, rt, pre = 'EAGNINQSLLTLGR', 2, 1, 2626.65, 743.40460205
pro = [446.27215576, 559.35620117, 672.44030762, 759.47229004, 887.53088379, 1001.5737915]
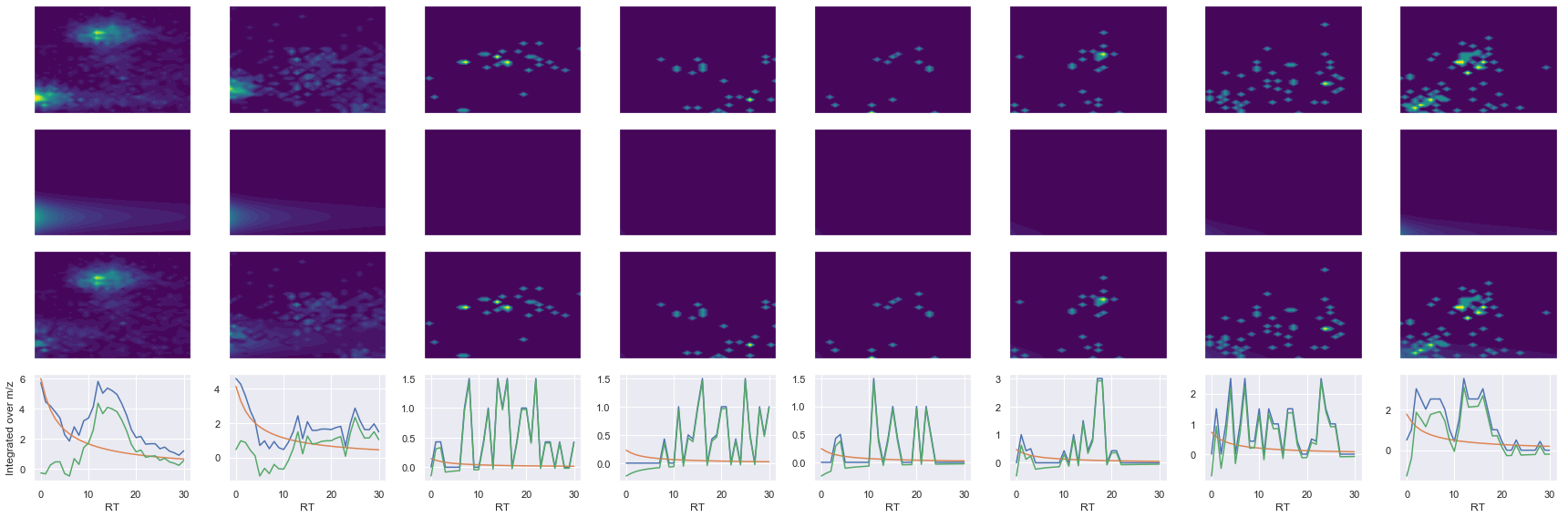
Raw MS1 & MS2: 5431.12478549956 124.70365258035318
Fit MS1 & MS2: 3157.8429424718797 36.32079911217963
[('EAGNINQSLLTLGR', 2, 3157.8429424718797, 36.32079911217963)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-089'
peptide, charge, rank, rt, pre = 'VEEASPGRPSSVDTLLSPTALIDSILR', 3, 1, 4217.8, 941.84143066
pro = [488.31912231, 603.34606934, 716.43011475, 1098.6517334, 1185.6837158, 1311.6175537]
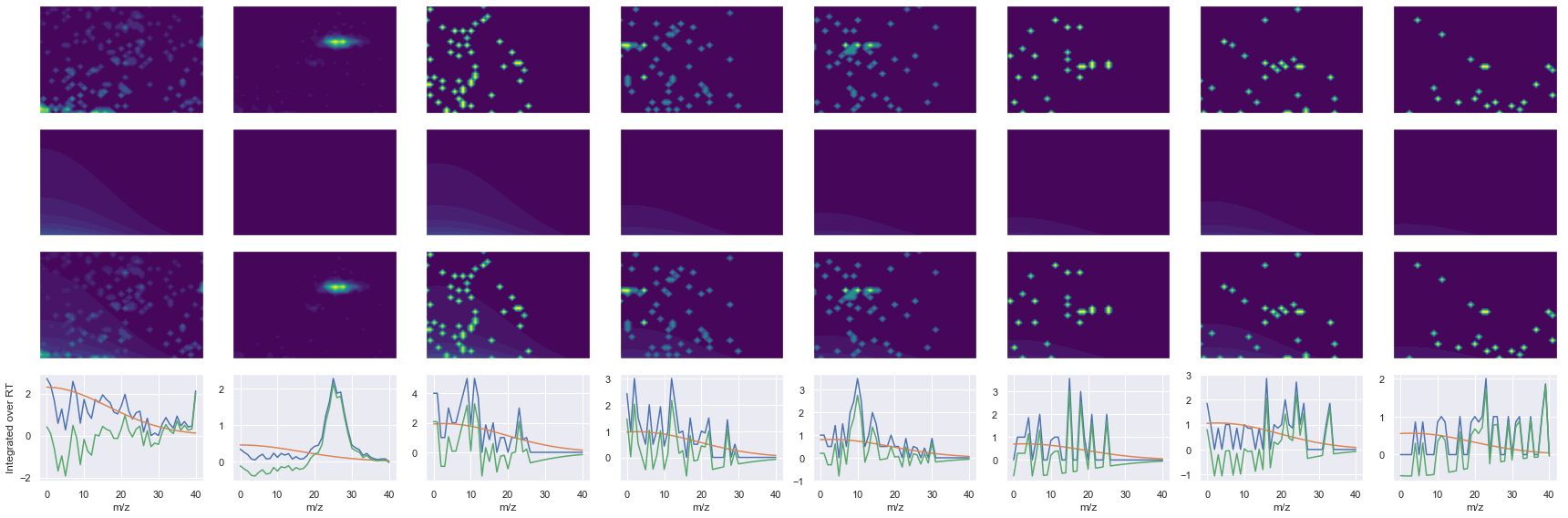
Raw MS1 & MS2: 2300.6600852753154 130.07829467804535
Fit MS1 & MS2: 699.0792733909628 98.30380772826076
[('VEEASPGRPSSVDTLLSPTALIDSILR', 3, 699.0792733909628, 98.30380772826076)]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-096'
peptide, charge, rank, rt, pre = 'SLLSIPNTDYIQLLSEIAK', 2, 1, 4206.23, 1059.5882568
pro = [660.39263916, 773.47674561, 901.53533936, 1177.6827393, 1393.7573242, 1604.8530273]
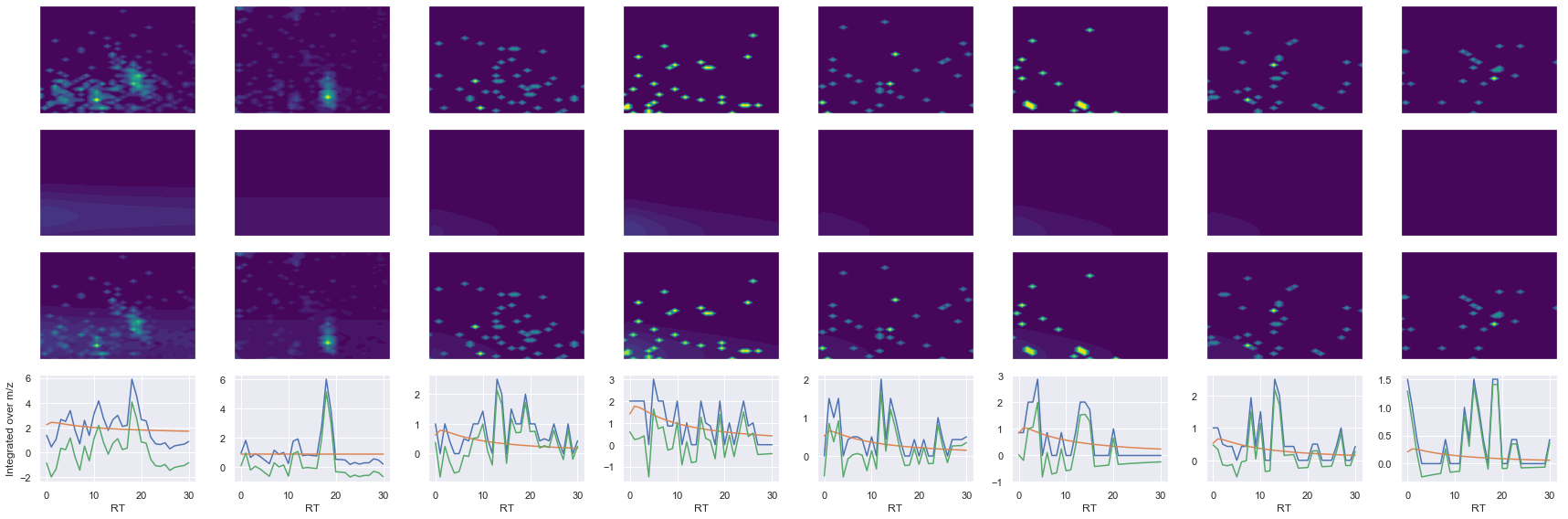
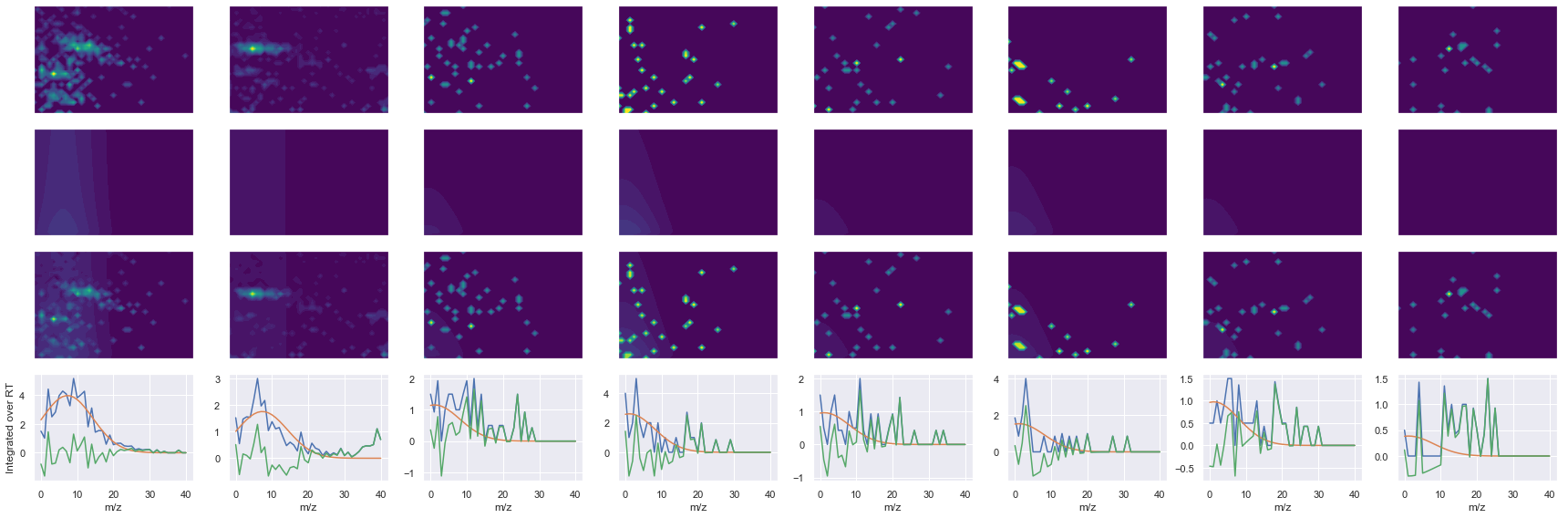
Raw MS1 & MS2: 2132.882937447126 114.49477907659808
Fit MS1 & MS2: 228.34049788928704 70.15997919249725
[('SLLSIPNTDYIQLLSEIAK', 2, 228.34049788928704, 70.15997919249725)]
Build testing data for the C++ library¶
We already use data from the DIA_TEST_DATA_REPO in our unit testing, so use the python implementation to build test data for the C++ tests.
[21]:
peptides_charges_and_rt = [
('DAVTPADFSEWSK', 2, 2609.69),
('EATFGVDESANK', 2, 830.17),
('GDLDAASYYAPVR', 2, 1882.61),
# ms2-025
('ELHINLIPNKQDR', 3, 1842.77),
('KSDIDEIVLVGGSTR', 3, 2231.21),
# ms2-052
('LAAEESVGTMGNR', 2, 826.85),
('TPVISGGPYYER', 2, 1467.61),
# ms2-056
('TLEEDEEELFK', 2, 2659.49),
('VQVALEELQDLK', 2, 3044.61),
# ms2-066
('GILAAEESVGTMGNR', 2, 1962.29),
('SPFTVGVAAPLDLSK', 2, 3263.73),
]
peptides = [p[0] for p in peptides_charges_and_rt]
srl_fname = base_dir + '/ProCan90/srl/hek_srl.OpenSwath.iRT.tsv'
tof_fname = base_dir + '/ProCan90/tof/ProCan90-M03-01.iRT.tof'
[22]:
plotter = ToffeeFragmentsPlotter(tof_fname)
[23]:
srl = pd.read_csv(srl_fname, sep='\t')
srl = srl.loc[srl.PeptideSequence.isin(peptides)]
ms2_map = plotter.extractor.swath_run.mapPrecursorsToMS2Names(srl.PrecursorMz.unique(), 0.0, 1.0)
srl['MS2Name'] = srl.PrecursorMz.map(ms2_map)
n_isotopes = 0
precursors, products = calculate_isotope_mz(
precursor_and_product_df=srl,
n_isotopes=n_isotopes,
sort_by_intensity=True,
)
all_srl = srl.copy()
for ms2_name in sorted(all_srl.MS2Name.unique()):
print('*' * 80)
srl = all_srl.loc[all_srl.MS2Name == ms2_name]
for peptide, charge, rt in [p for p in peptides_charges_and_rt if p[0] in srl.PeptideSequence.unique()]:
print('-' * 50)
pqp = srl.loc[srl.PeptideSequence == peptide, 'TransitionGroupId'].unique()[0]
ms2_name = srl.loc[srl.PeptideSequence == peptide, 'MS2Name'].unique()[0]
pre = precursors.loc[precursors.Id == pqp, 'IsotopeMz'].tolist()
pro = products.loc[products.GroupId == pqp, 'IsotopeMz'].tolist()[:6]
assert len(pre) > 0
assert len(pro) > 0
print(f'ms2_name = \'{ms2_name}\'')
print(f'peptide, charge, rt, pre = \'{peptide}\', {charge}, {rt}, {pre[0]}')
print(f'pro = {pro}')
output_fname = '/tmp/fig.png'
fig = plotter.create_plotly_figure(
precursor_mz_list=pre,
product_mz_list=pro,
psm_name='{} | extraction_width = {} {}'.format(
plotter.tof_fname.split('/')[-1].split('.')[0],
plotter.extractor.extraction_width,
'ppm' if plotter.extractor.use_ppm else 'px',
),
number_of_isotopes=n_isotopes,
log_heatmap=True,
normalise_per_fragment=False,
retention_time_line=rt,
output_fname=output_fname,
)
display(Image(output_fname))
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-025'
peptide, charge, rt, pre = 'ELHINLIPNKQDR', 3, 1842.77, 530.630126953125
pro = [757.395141601563, 607.31982421875, 720.403930664063, 660.342346191406, 546.2994384765631, 1097.60620117188]
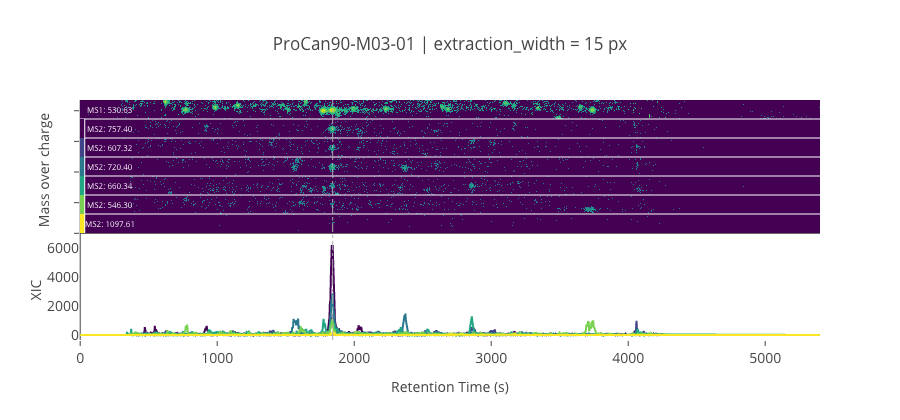
--------------------------------------------------
ms2_name = 'ms2-025'
peptide, charge, rt, pre = 'KSDIDEIVLVGGSTR', 3, 2231.21, 530.28955078125
pro = [576.309997558594, 801.3988647460941, 689.39404296875, 688.314819335938, 900.46728515625, 559.272216796875]
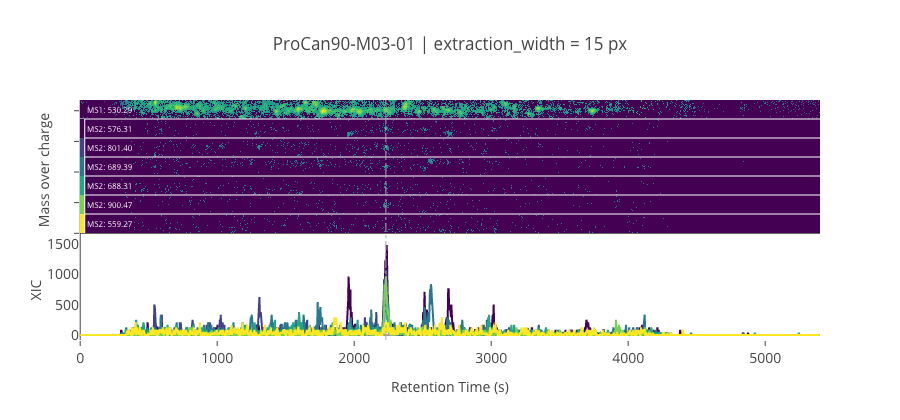
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-046'
peptide, charge, rt, pre = 'EATFGVDESANK', 2, 830.17, 634.293701171875
pro = [819.38427734375, 663.29443359375, 419.22488403320295, 966.4526977539059, 548.2674560546881, 762.3628540039059]
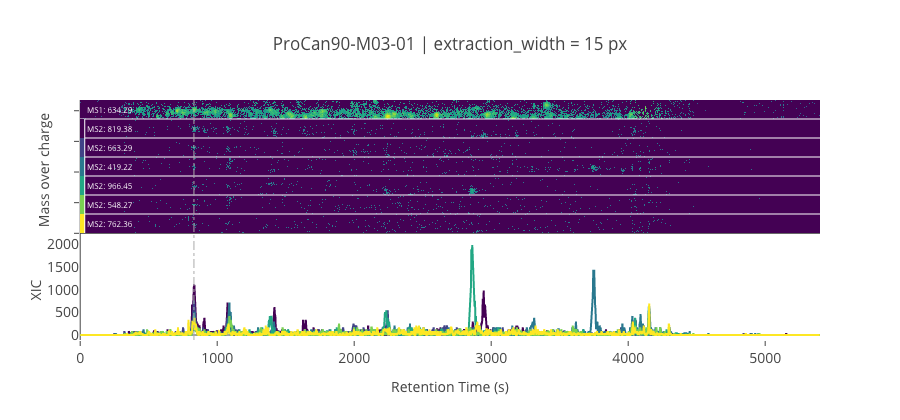
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-052'
peptide, charge, rt, pre = 'LAAEESVGTMGNR', 2, 826.85, 667.822082519531
pro = [635.29296875, 821.393432617188, 950.43603515625, 1150.51574707031, 734.3613891601559, 1079.47863769531]
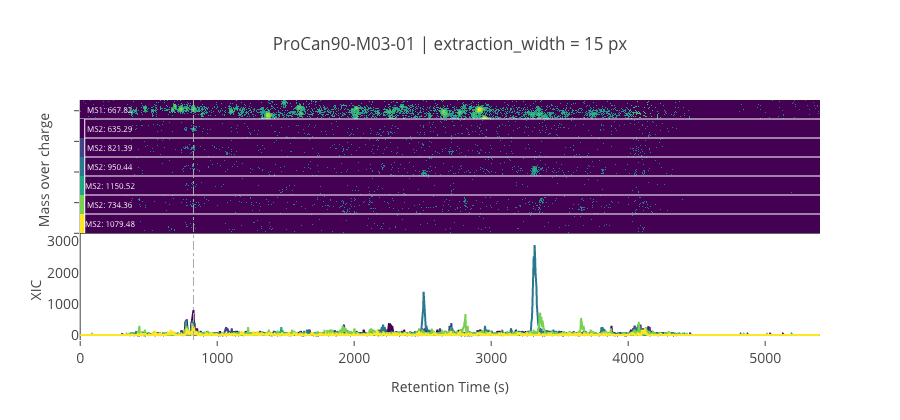
--------------------------------------------------
ms2_name = 'ms2-052'
peptide, charge, rt, pre = 'TPVISGGPYYER', 2, 1467.61, 669.838073730469
pro = [928.415893554688, 1041.5, 1140.568359375, 841.383911132813, 784.362426757813, 727.340942382813]
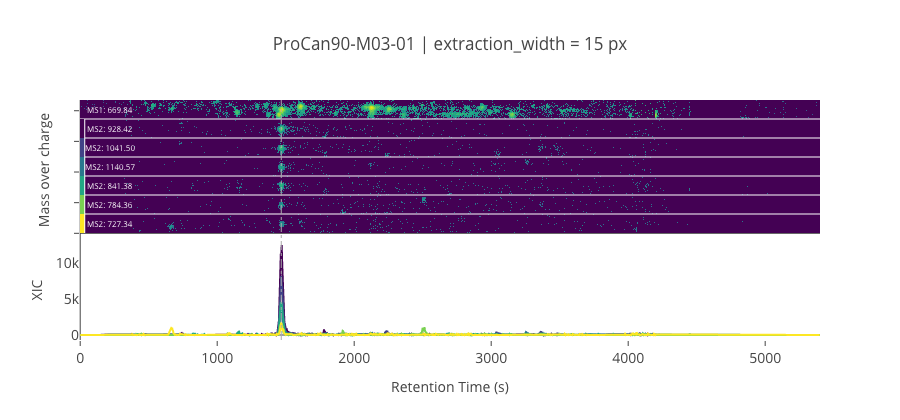
********************************************************************************
--------------------------------------------------
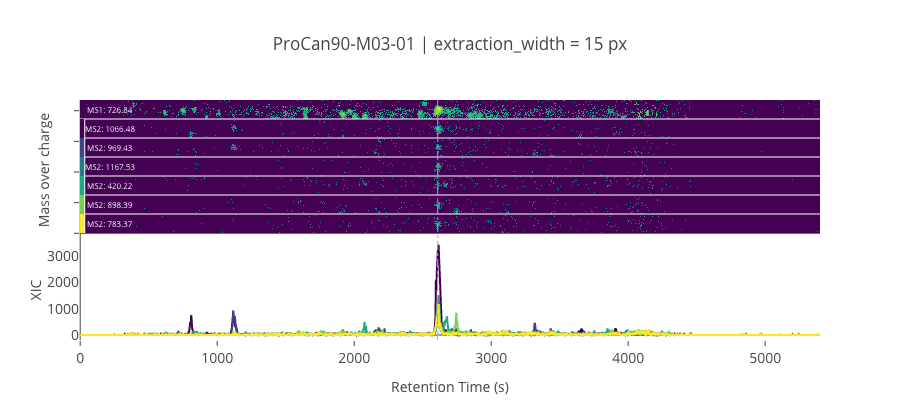
ms2_name = 'ms2-056'
peptide, charge, rt, pre = 'TLEEDEEELFK', 2, 2659.49, 691.3220825195309
pro = [1167.50524902344, 1038.46264648438, 909.4199829101559, 794.39306640625, 717.2937622070309, 846.3363647460941]
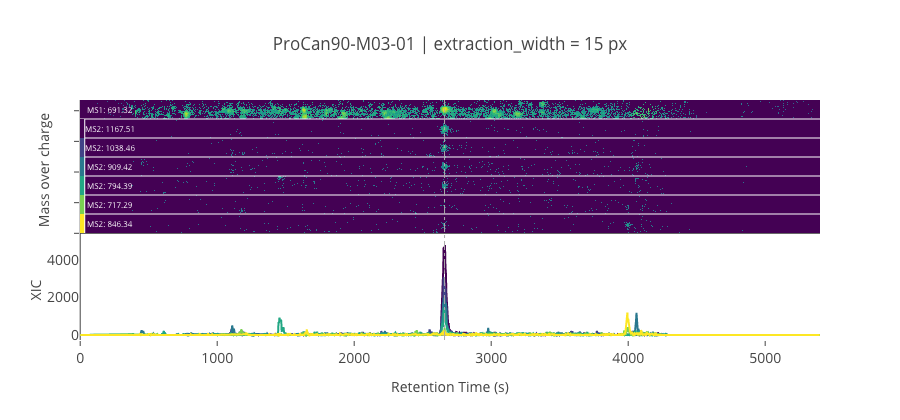
--------------------------------------------------
ms2_name = 'ms2-056'
peptide, charge, rt, pre = 'VQVALEELQDLK', 2, 3044.61, 692.8877563476559
pro = [1058.57287597656, 1157.64123535156, 987.5357055664059, 616.366455078125, 874.45166015625, 745.409057617188]
********************************************************************************
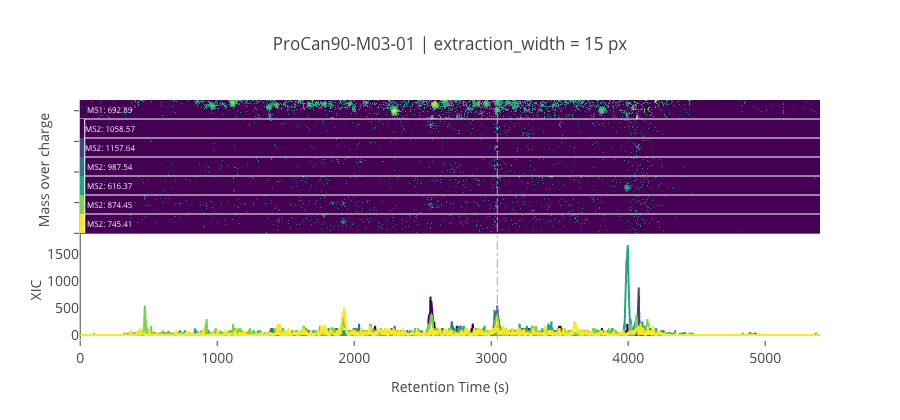
--------------------------------------------------
ms2_name = 'ms2-057'
peptide, charge, rt, pre = 'GDLDAASYYAPVR', 2, 1882.61, 699.3384399414059
pro = [855.435913085938, 926.473022460938, 997.510131835938, 768.403930664063, 605.340576171875, 1112.537109375]
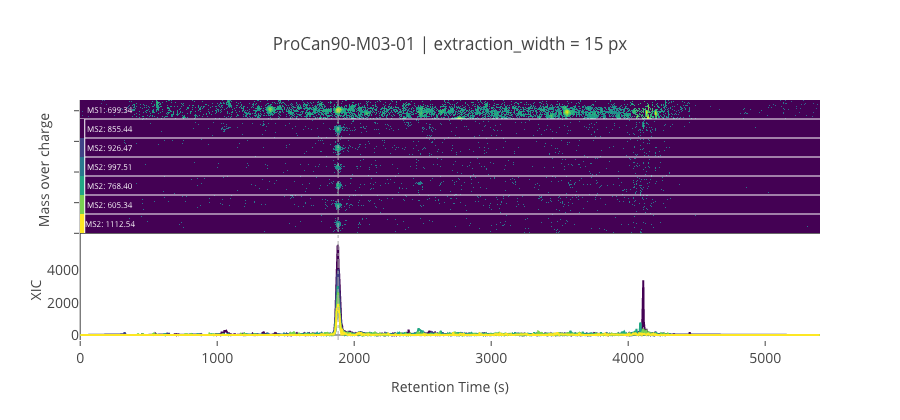
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-062'
peptide, charge, rt, pre = 'DAVTPADFSEWSK', 2, 2609.69, 726.835693359375
pro = [1066.48400878906, 969.4312133789059, 1167.53173828125, 420.22415161132795, 898.3941040039059, 783.3671875]
********************************************************************************
--------------------------------------------------
ms2_name = 'ms2-066'
peptide, charge, rt, pre = 'GILAAEESVGTMGNR', 2, 1962.29, 752.8748168945309
pro = [1150.51574707031, 1221.55285644531, 635.29296875, 821.393432617188, 1079.47863769531, 950.43603515625]
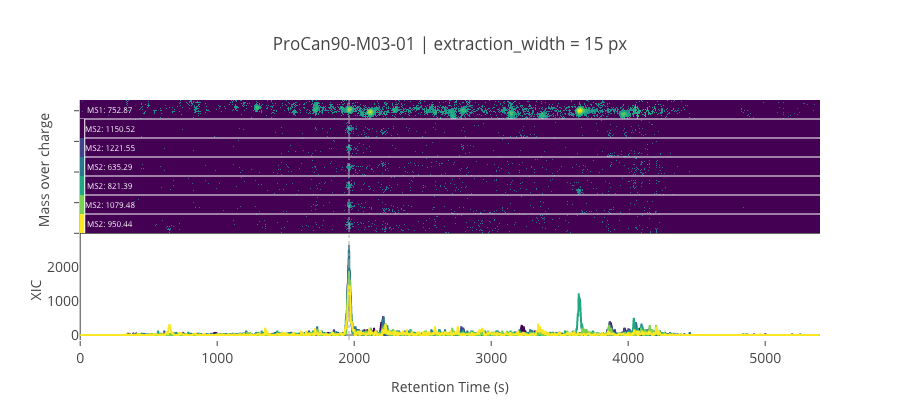
--------------------------------------------------
ms2_name = 'ms2-066'
peptide, charge, rt, pre = 'SPFTVGVAAPLDLSK', 2, 3263.73, 751.4166870117191
pro = [970.556762695313, 814.466918945313, 1069.62524414063, 1170.6728515625, 913.5353393554691, 1317.74133300781]
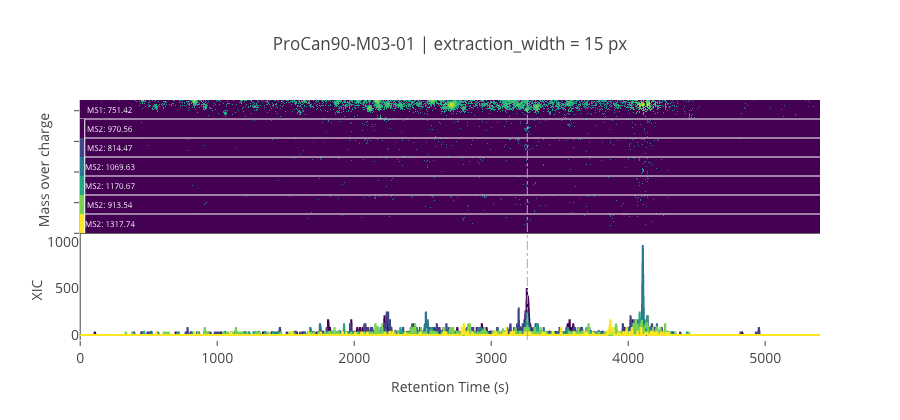
[24]:
operator = Requant(tof_fname, debug_plots=True)
for ms2_name in sorted(all_srl.MS2Name.unique()):
print('*' * 80)
srl = all_srl.loc[all_srl.MS2Name == ms2_name]
for peptide, charge, rt in [p for p in peptides_charges_and_rt if p[0] in srl.PeptideSequence.unique()]:
print('-' * 50)
pqp = srl.loc[srl.PeptideSequence == peptide, 'TransitionGroupId'].unique()[0]
charge = srl.loc[srl.PeptideSequence == peptide, 'PrecursorCharge'].unique()[0]
ms2_name = srl.loc[srl.PeptideSequence == peptide, 'MS2Name'].unique()[0]
rank = 1
pre = precursors.loc[precursors.Id == pqp, 'IsotopeMz'].tolist()
pro = products.loc[products.GroupId == pqp, 'IsotopeMz'].tolist()[:6]
assert len(pre) > 0
assert len(pro) > 0
print(pqp, ms2_name)
peak_group = requant.RequantPeakGroup(peptide, charge, rank, rt, pre[0], pro)
print(operator.run(ms2_name, [peak_group]))
********************************************************************************
--------------------------------------------------
ELHINLIPNKQDR_3 ms2-025
546.2994384765631 (31, 38) != (31, 41)
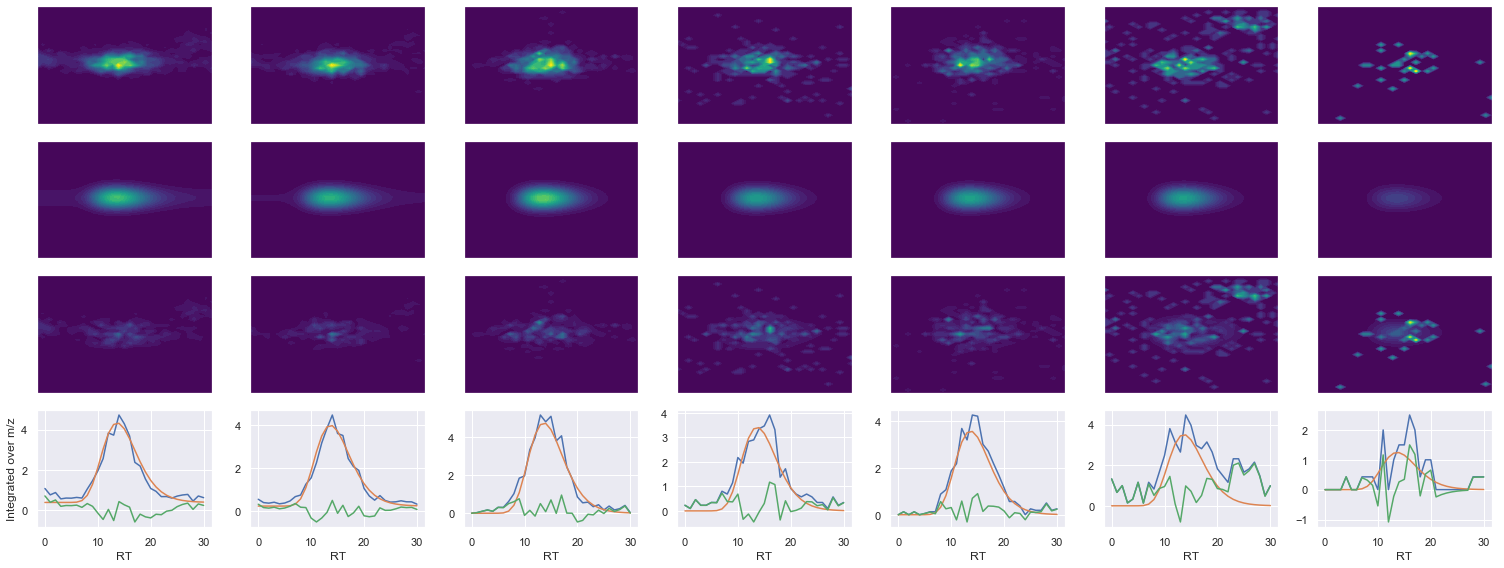
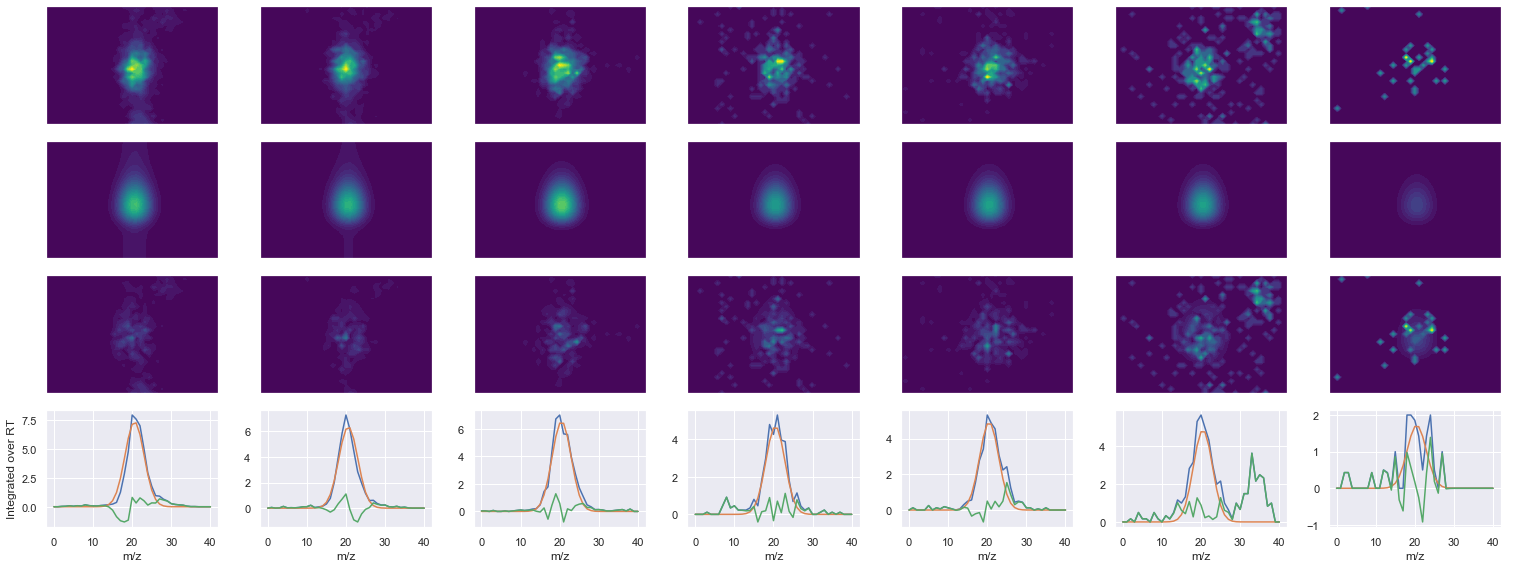
Raw MS1 & MS2: 7971.382612753305 1360.6360110167914
Fit MS1 & MS2: 5936.0447804902815 1105.2600737423677
[('ELHINLIPNKQDR', 3, 5936.0447804902815, 1105.2600737423677)]
--------------------------------------------------
KSDIDEIVLVGGSTR_3 ms2-025
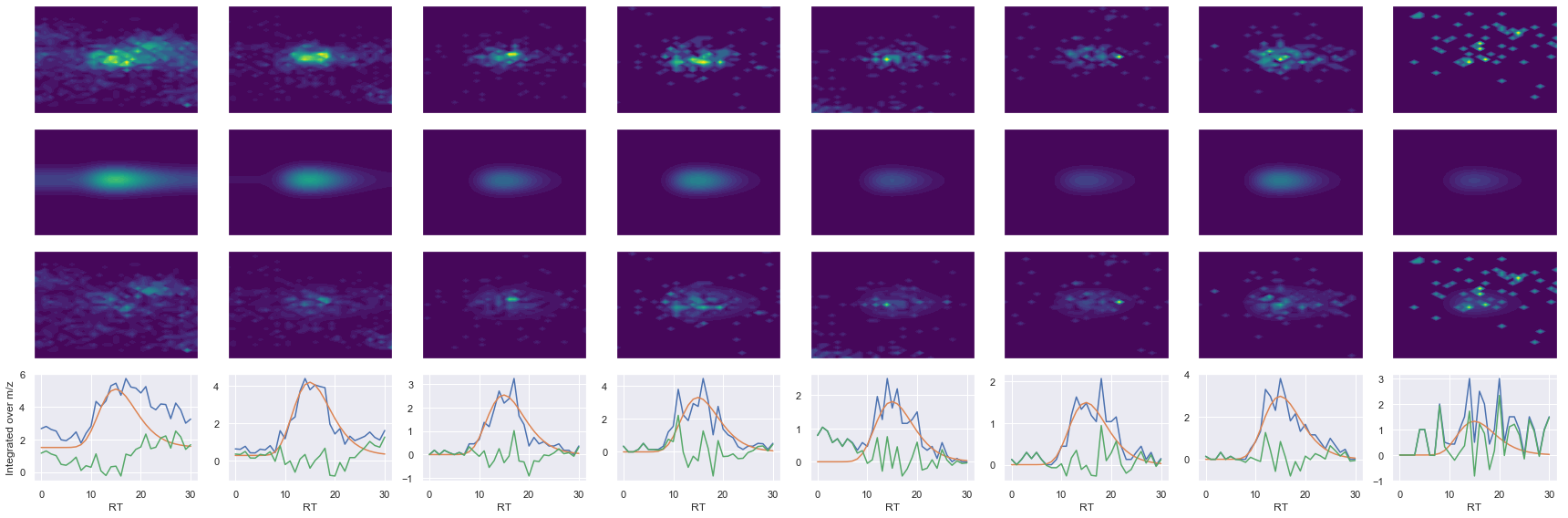
Raw MS1 & MS2: 3663.5420920012957 548.9509062646167
Fit MS1 & MS2: 1565.341063713942 467.98236860566055
[('KSDIDEIVLVGGSTR', 3, 1565.341063713942, 467.98236860566055)]
********************************************************************************
--------------------------------------------------
EATFGVDESANK_2 ms2-046
419.22488403320295 (31, 36) != (31, 41)
Raw MS1 & MS2: 2727.821488812674 294.9427344606671
Fit MS1 & MS2: 1408.4605688391075 249.52289699122136
[('EATFGVDESANK', 2, 1408.4605688391075, 249.52289699122136)]
********************************************************************************
--------------------------------------------------
LAAEESVGTMGNR_2 ms2-052
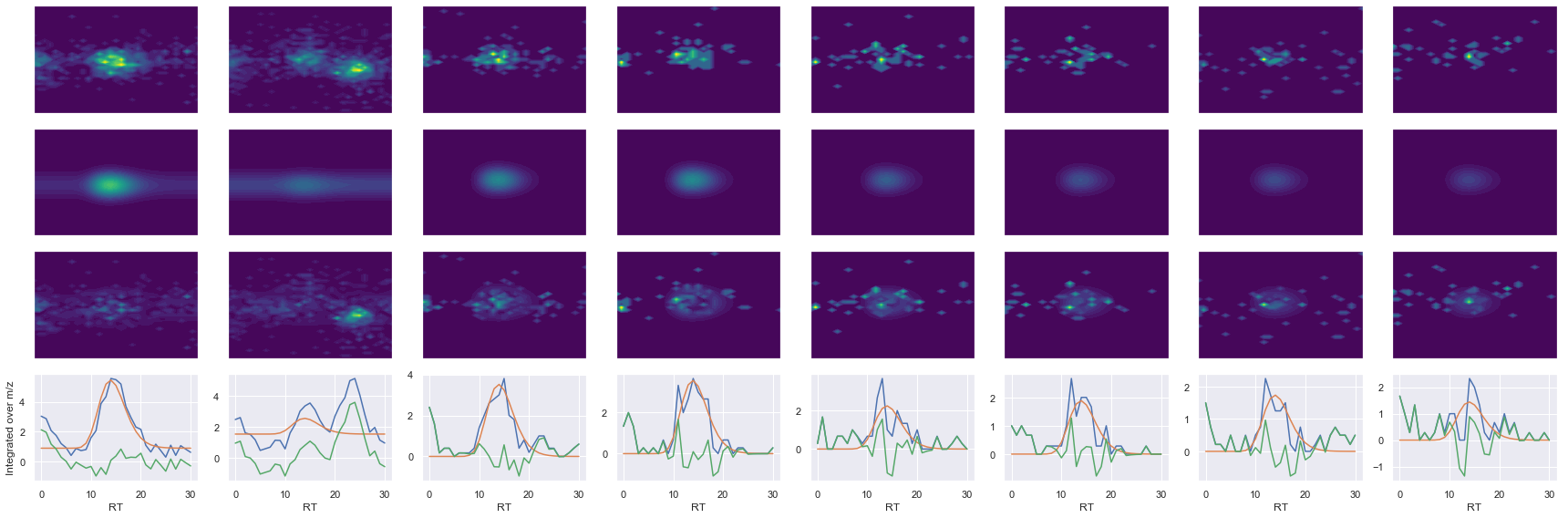
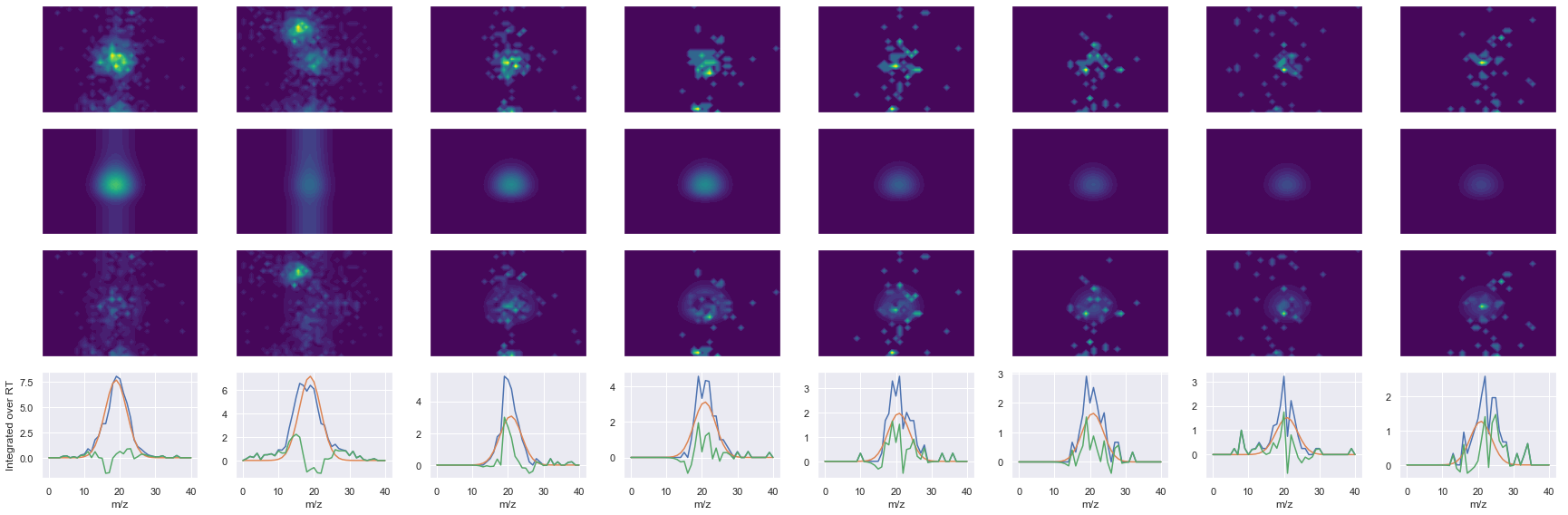
Raw MS1 & MS2: 2402.871796360551 274.4929809051114
Fit MS1 & MS2: 640.5041539081958 193.0167084570027
[('LAAEESVGTMGNR', 2, 640.5041539081958, 193.0167084570027)]
--------------------------------------------------
TPVISGGPYYER_2 ms2-052
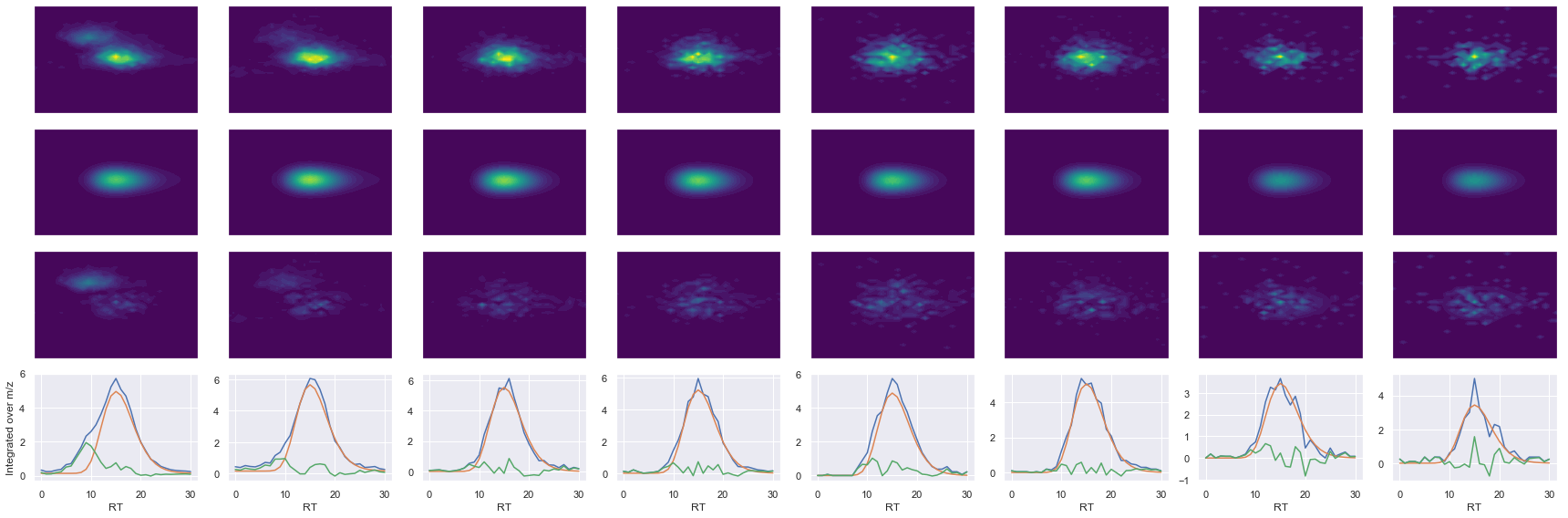
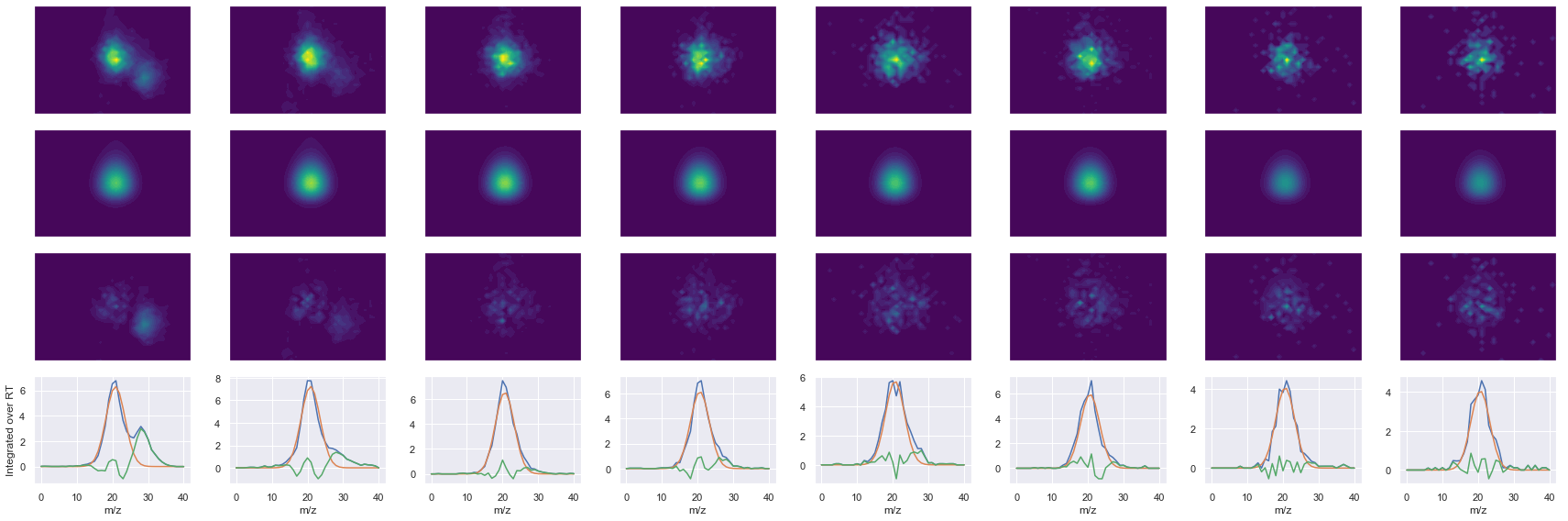
Raw MS1 & MS2: 18987.131273955467 3611.313791924786
Fit MS1 & MS2: 13335.611364021508 3232.3448694321823
[('TPVISGGPYYER', 2, 13335.611364021508, 3232.3448694321823)]
********************************************************************************
--------------------------------------------------
TLEEDEEELFK_2 ms2-056
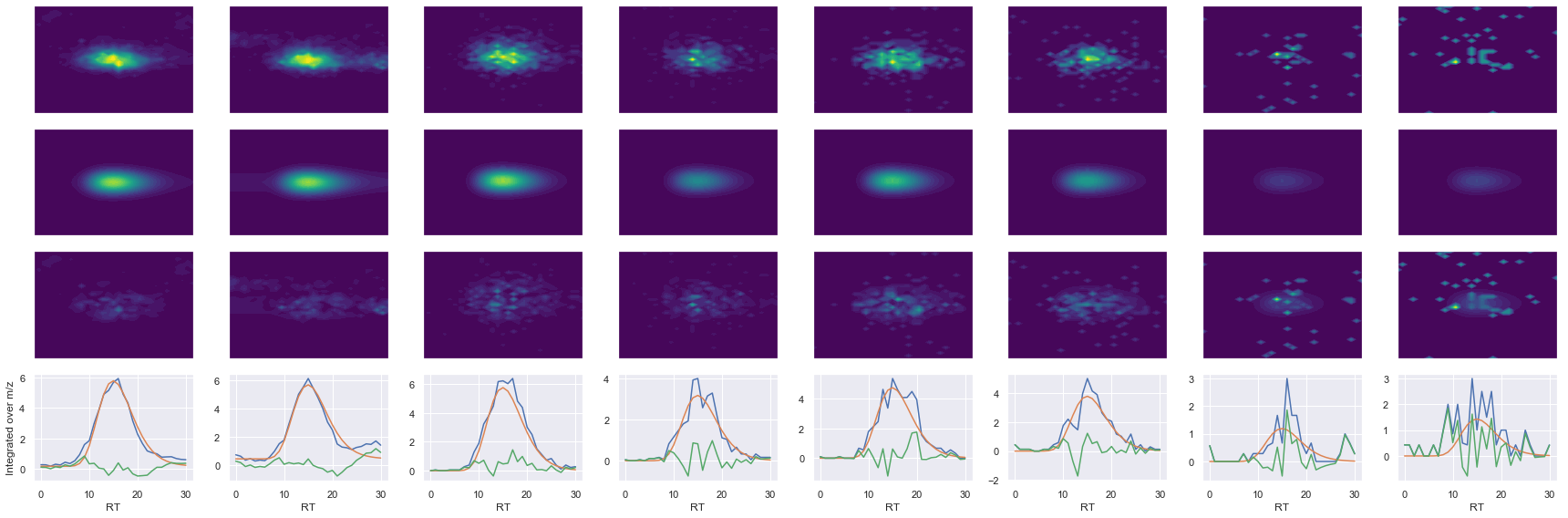
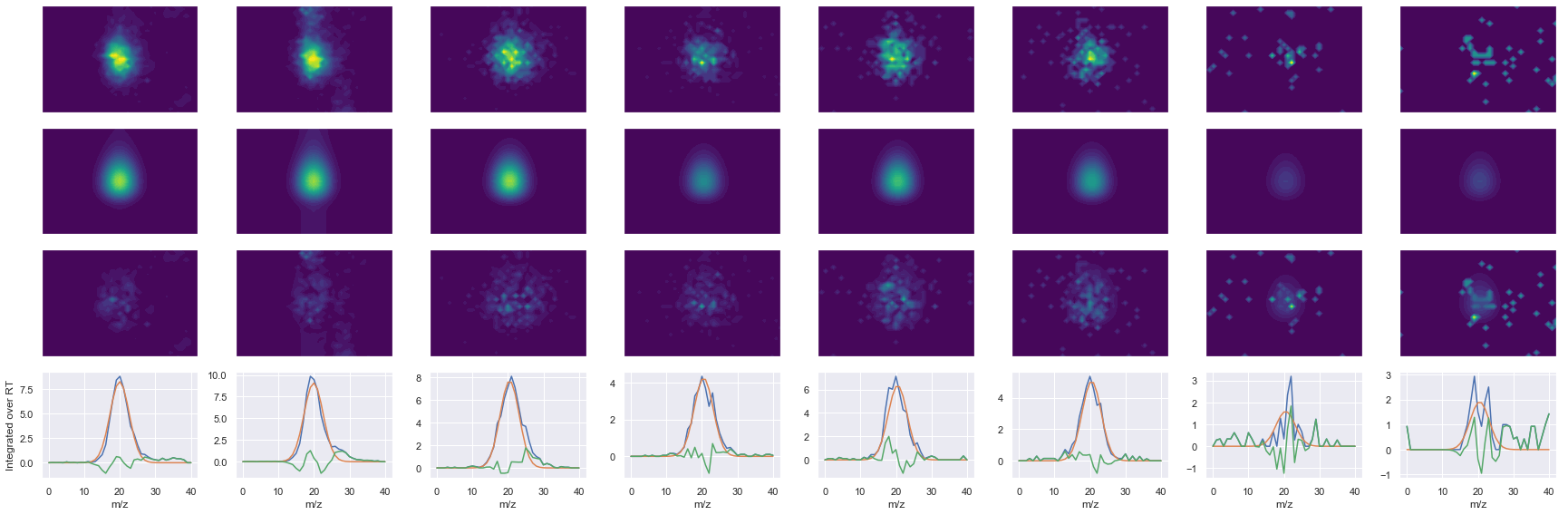
Raw MS1 & MS2: 14489.968375957485 1537.2048833212073
Fit MS1 & MS2: 11542.81942725199 1334.0724131818538
[('TLEEDEEELFK', 2, 11542.81942725199, 1334.0724131818538)]
--------------------------------------------------
VQVALEELQDLK_2 ms2-056
616.366455078125 (31, 40) != (31, 41)
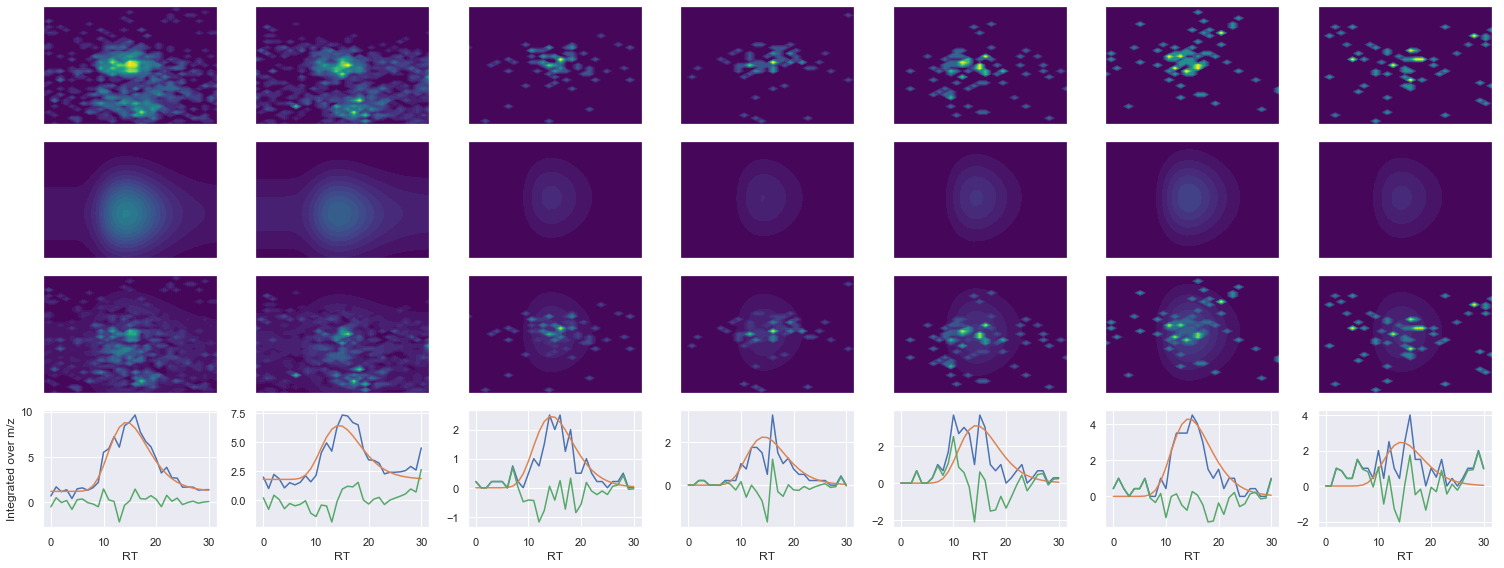
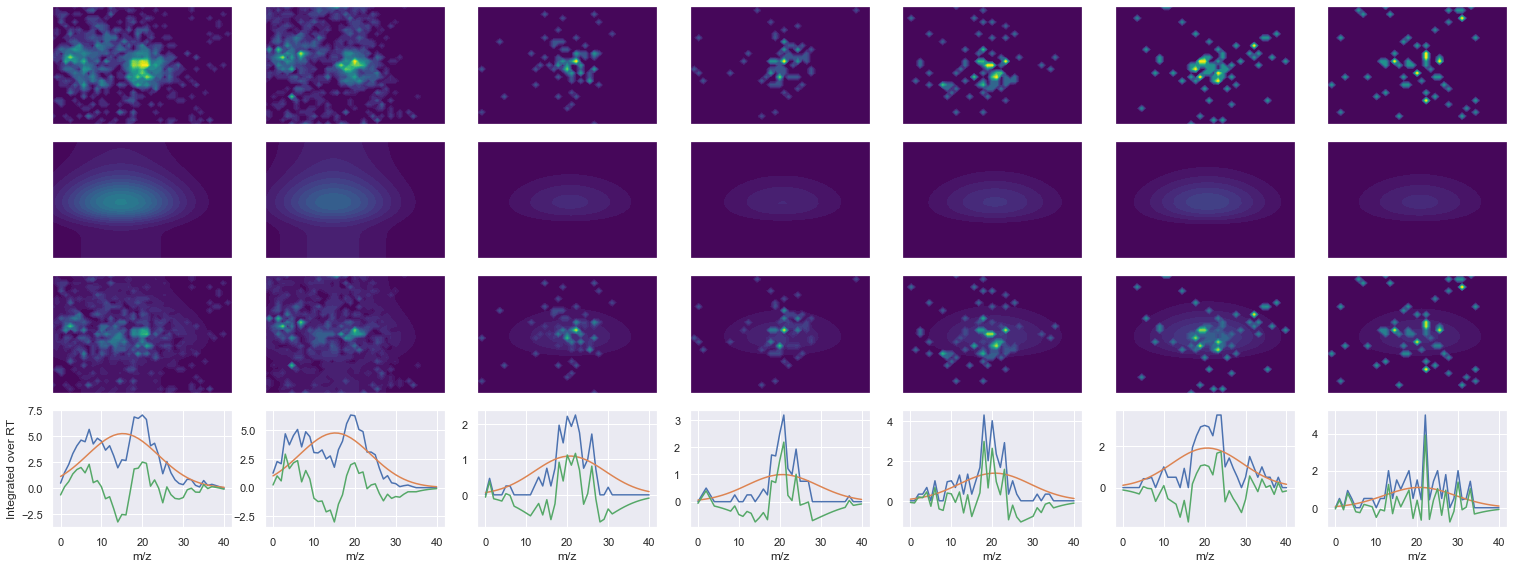
Raw MS1 & MS2: 2704.855085515457 233.11135740071734
Fit MS1 & MS2: 1448.0969122618142 242.93041021698042
[('VQVALEELQDLK', 2, 1448.0969122618142, 242.93041021698042)]
********************************************************************************
--------------------------------------------------
GDLDAASYYAPVR_2 ms2-057
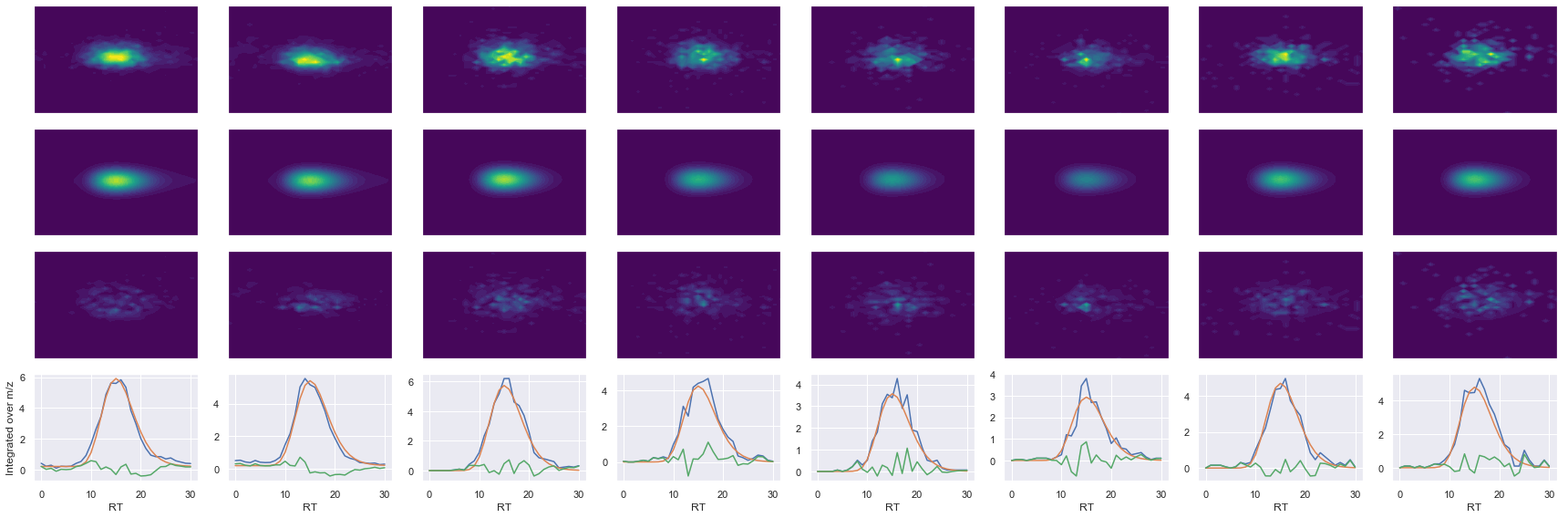
Raw MS1 & MS2: 9132.382213427156 2131.9018901521886
Fit MS1 & MS2: 7664.530453314555 1927.7800850246947
[('GDLDAASYYAPVR', 2, 7664.530453314555, 1927.7800850246947)]
********************************************************************************
--------------------------------------------------
DAVTPADFSEWSK_2 ms2-062
420.22415161132795 (31, 36) != (31, 41)
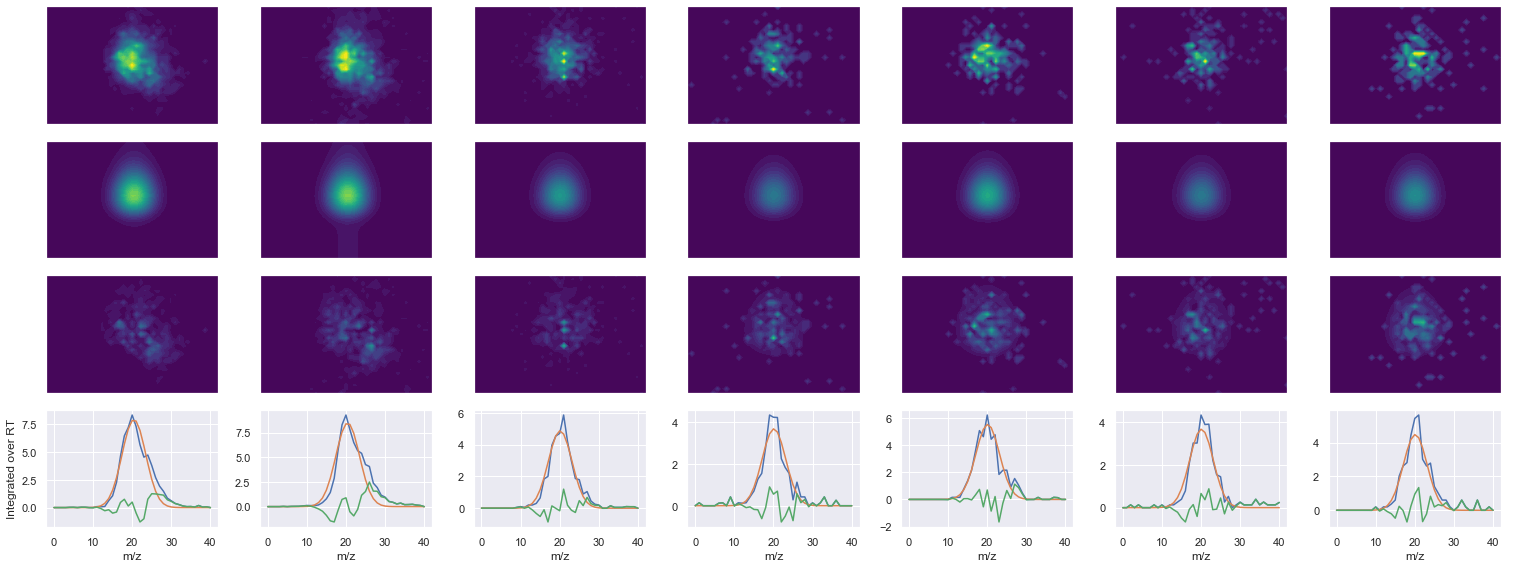
Raw MS1 & MS2: 10443.635559962993 1055.7539806959778
Fit MS1 & MS2: 8230.962938048931 991.0828664359409
[('DAVTPADFSEWSK', 2, 8230.962938048931, 991.0828664359409)]
********************************************************************************
--------------------------------------------------
GILAAEESVGTMGNR_2 ms2-066
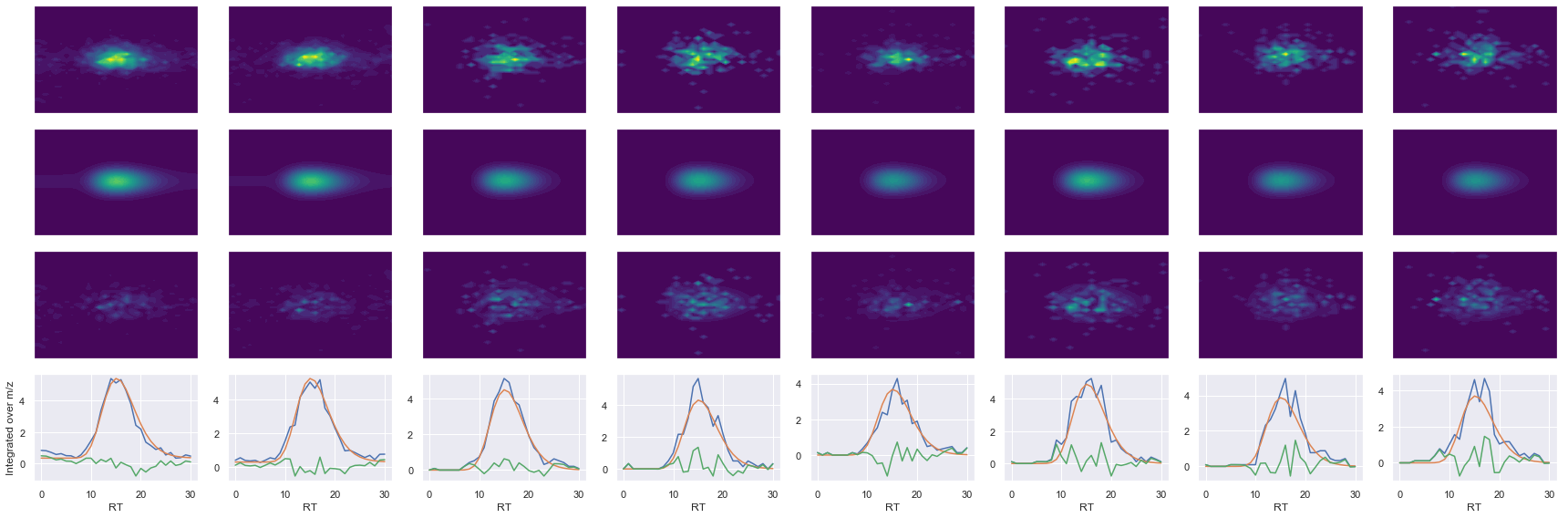
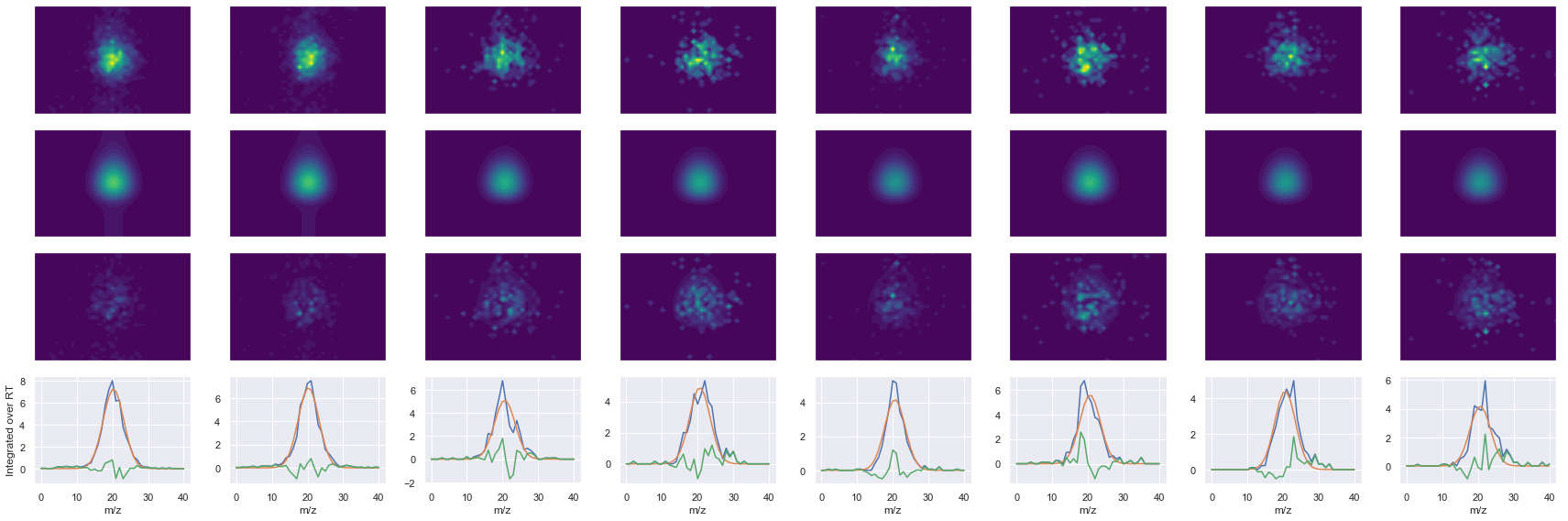
Raw MS1 & MS2: 6142.502002452005 1221.8592417017833
Fit MS1 & MS2: 4804.03914396974 1101.5187370787942
[('GILAAEESVGTMGNR', 2, 4804.03914396974, 1101.5187370787942)]
--------------------------------------------------
SPFTVGVAAPLDLSK_2 ms2-066
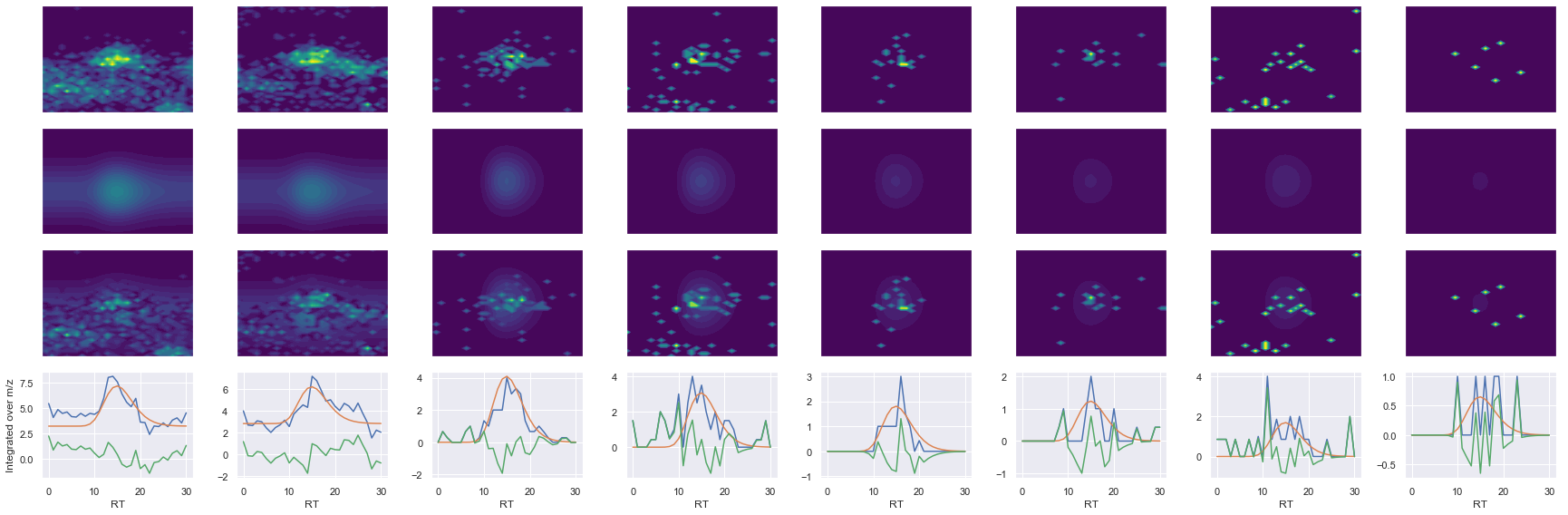
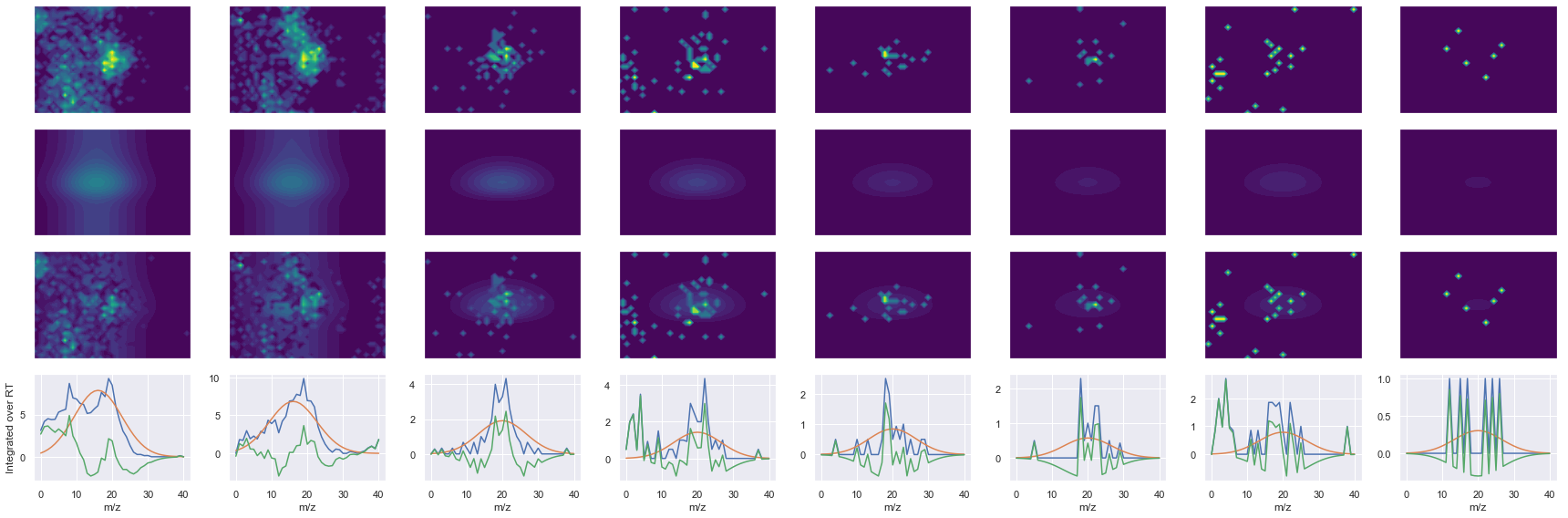
Raw MS1 & MS2: 2656.414106639162 136.0974727587502
Fit MS1 & MS2: 580.3664099675456 126.41985619771064
[('SPFTVGVAAPLDLSK', 2, 580.3664099675456, 126.41985619771064)]
[ ]: